Acute clinical-histopathologic patterns
Typical drugs
Diffuse alveolar damage (DAD/ARDS)
Bleomycin, busulfan, cyclophosphamide, mitomycin, amiodarone
Diffuse alveolar hemorrhage (DAH)
Anticoagulants, amphotericin B, cytarabine (ara-C), penicillamine, cyclophosphamide
Pulmonary edema (PE)
Blood transfusions, tricyclic antidepressants, illicit drugs
Hypersensitivity pneumonia (HP)
Methotrexate, cyclophosphamide, nitrofurantoin
1.5 Management and Treatment
Disease types such as pulmonary edema and hypersensitivity pneumonia generally have a favorable clinical course and most patients resolve following drug discontinuation or treatment with corticosteroids. In contrast, DAD rarely responds to treatment and has a poor prognosis, and even if it resolves, fibrosis may remain as a sequela.
2 Hypersensitivity Pneumonitis (HP)
2.1 Introduction
Hypersensitivity pneumonitis (HP), also known as extrinsic allergic alveolitis, is a syndrome that results from repeated inhalation and subsequent sensitization to a wide variety of airborne organic particles. The presentation and clinical course are highly variable and depend from intensity and duration of exposure to the antigen and the nature of the antigen and specific factors of the host, such as an individual predisposition probably genetically determined (Spagnolo et al. 2015). According to data from registries of interstitial lung diseases (ILDs) in three European countries, HP accounts for 4–15 % of all ILD cases (Thomeer et al. 2001). However, the incidence and prevalence of HP are difficult to estimate with precision, mainly because of the number of cases that are misdiagnosed or not recognized and a lack of uniform diagnostic criteria.
The disease is diagnosed on the basis of a history of exposure to an offending antigen with onset of compatible clinical, HRCT, or physiological findings within 4–12 h. Other diagnostic criteria include clinical improvement after removal from exposure and recurrence on reexposure. In cases where the inciting antigen cannot be identified or in the presence of conflicting clinical, radiological, and functional findings, fiberoptic bronchoscopy with bronchoalveolar lavage (BAL) and transbronchial lung biopsy are indicated. Surgical lung biopsy is only required if these prove inconclusive (Elicker et al. 2016).
2.2 Mechanisms of Injury and Causes
HP comes from an immune-mediated inflammatory process involving the lung parenchyma (terminal bronchioles, alveoli, and interstitium), based on mechanisms likely independent from single causative agent, mediated by immune complexes in the acute phases of the disease and an altered response of T lymphocytes in the early stages subacute and chronic (Vogelmeier et al. 1993; Barrera et al. 2008). HP seems that cigarette smoking can play a protective effect by reducing the intensity of hyperimmune response. The immune pathogenesis, not yet fully clarified, is linked to hyperactivity of T cells and the action of immune complexes.
In every living environment, antigens potentially causative of the disease may be present. The reported culprits have included microbes, animal and plant proteins, and low-molecular-weight chemicals that combine with host proteins to form haptens. HP-inducing antigens may be classified in broad categories represented by disease prototypes (Table 2) (Hanak et al. 2007; Spagnolo et al. 2015; Selman 2011). In the table is a selected list of the possible innumerable agents and disease.
Table 2
Major classes of antigens and corresponding types of hypersensitivity pneumonitis
Class of antigens | Specific antigens | Disease prototypes |
|---|---|---|
Fungi | Aspergillus | Farmer’s lung |
Mushrooms worker’s lung | ||
Mycobacteria | Mycobacterium avium–intracellulare | Hot tub lung |
Swimming pool lung | ||
Bacteria | Saccharopolyspora rectivirgula | Farmer’s lung |
Animal proteins | Avian proteins | Bird fancier’s lung |
Silkworm proteins | Silk production HP | |
Chemicals | Diisocyanates, trimellitic anhydride | Chemical worker’s lung |
2.3 Terminology and Clinical Issues
HP has been conventionally classified as acute, subacute, and chronic (Richerson et al. 1989), although a significant overlap exists and there are no widely accepted criteria to distinguish the various forms. In this regard, (Selman et al. 2012) proposed an alternative classification scheme based primarily on disease behavior, distinguishing between acute nonprogressive and intermittent disease, acute progressive/subacute disease, chronic nonprogressive disease, and chronic progressive disease. Which pattern of illness occurs presumably depends upon the intensity and duration of contact, the nature of the antigen, and host factors.
Acute HP is characterized by an influenza-like syndrome occurring a few hours after a substantial exposure. Symptoms gradually decrease over hours/days but often recur with reexposure. Acute episodes can be indistinguishable from an acute respiratory infection caused by viral or mycoplasmal agents. Attacks often follow exposure to the allergen within enclosed spaces with poor ventilation. Patients typically have a restrictive ventilatory defect with reduced DLCO or, in rare cases, an obstructive pattern. Mild hypoxemia at rest is common. In general, the acute form is nonprogressive and intermittent, with spontaneous improvement after antigen avoidance (Selman et al. 2012).
2.4 Imaging
2.4.1 Radiological Signs
Chest radiograph may be normal. Abnormal radiographic findings observed in some patients include a variable combination of fine nodular opacities and widespread ground-glass opacity or, more rarely, as consolidation. The zonal distribution varies from patient to patient and may vary over time in the same patient (Unger et al. 1973; Mönkäre et al. 1985).
2.4.2 CT Signs
High-resolution CT has greatly improved the radiological diagnosis of hypersensitivity pneumonitis because it is more sensitive and specific than chest X-ray. HRCT may either show typical findings, which may be virtually diagnostic of HP in the appropriate clinical setting, or provide important clues that may suggest a correct diagnosis.
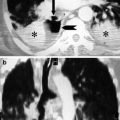
HRCT findings vary widely based on the stages of the disease. Only a few reports have described HRCT abnormalities in acute HP, as HRCT is seldom performed at this stage due to the rapid resolution of symptoms. However, in cases with severe clinical manifestations (e.g., acute respiratory failure), acute HP on HRCT scans may resemble the exudative phase of diffuse alveolar damage (DAD) (Schwarz and Albert 2004). Diffuse ground-glass opacity is usually bilateral and symmetric but sometimes patchy and concentrated in the middle part and base of the lungs (Cormier et al. 2000; Zompatori et al. 2003) (Fig. 1). Furthermore, ground-glass opacity (GGO) superimposed on a background of chronic changes may be observed in acute exacerbation of chronic HP or in chronic cases following intense exposure to antigens.

Fig. 1
Hypersensitivity pneumonitis with acute onset. High-resolution computed tomography showing diffuse ground-glass opacity, which is bilateral and symmetric
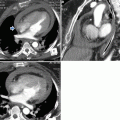
HRCT abnormalities observed in the subacute phase of the disease may be more specific and include numerous poorly defined nodules, GGO, and areas of decreased attenuation. The nodules with low-density and ill-defined margins usually present less than 5 mm in diameter (also defined centrilobular ground-glass opacities). In terms of their aspect, the nodules are similar to snowflakes. Poorly defined nodules may be the predominant (Fig. 2) or only associated abnormality in patients with subacute HP. These abnormalities represent cellular bronchiolitis, peribronchiolar interstitial inflammation, or, less frequently, focal organizing pneumonia (Maffessanti and Dalpaz 2011).
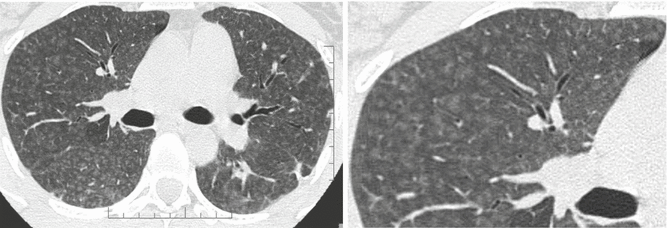
Fig. 2
Hypersensitivity pneumonitis. Axial high-resolution CT images depict bilateral and numerous centrilobular nodules with low-density and ill-defined margins. In terms of their aspect, the nodules are similar to snowflakes. The nodules are diffuse with uniform distribution. Dark areas of lobular size due to air trapping are also visible
The key sign to the diagnosis is the coexistence of sporadic lobular areas of air trapping appearing as patches of black lung (Fig. 3). These regions of lobular air trapping are caused by concomitant bronchiolar inflammation and obstruction.
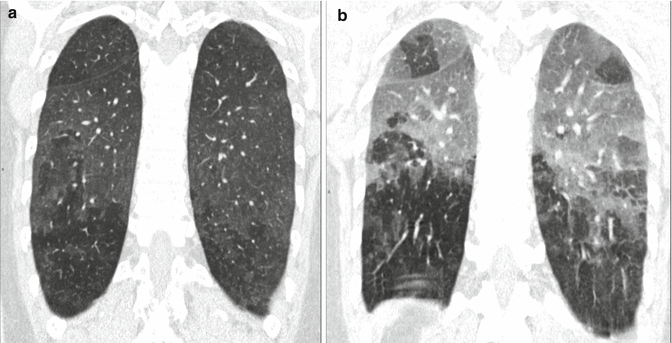
Fig. 3
Recurrent bird fancier’s disease. (a) Coronal reformatted computed tomography (CT) image obtained at suspended inspiration demonstrates patchy ground glass involving the medium-upper zones combined with areas of decreased attenuation. (b) The intensity of the areas of decreased attenuation increases on expiratory CT indicating small airway involvement, an almost invariable finding in hypersensitivity pneumonitis. A few dark areas of lobular size due to air trapping are visible in the upper lobes
Thus, the combination of a ground-glass opacification, lobular air trapping, and centrilobular nodules is particularly suggestive of acute and insidious onset without fibrosis (Fig. 4) (Silva et al. 2007).
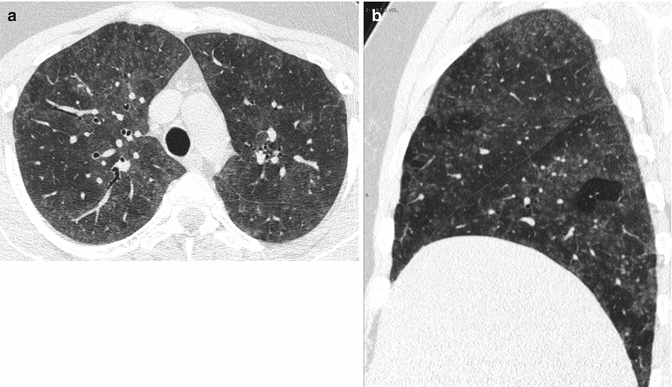
Fig. 4
(a, b) Typical high-resolution CT abnormalities in patients with hypersensitivity pneumonitis with insidious course and recurrent acute exacerbation include ground-glass opacity, air trapping, and centrilobular ground-glass opacities
The variable combination of areas of decreased attenuation, ground-glass opacities, and normal lung may produce the so-called head-cheese pattern, which is highly suggestive of HP (Chong et al. 2014). Coexisting thin-walled lung cysts have been reported in 13 % of patients with subacute HP (Franquet et al. 2003) and are believed to be caused by partial bronchiolar obstruction by peribronchiolar lymphocytic infiltration. These cysts are usually few in number and range in size from 3 to 25 mm. Occasionally in patients with an insidious onset of disease, focal consolidation is present, presumably representing organizing pneumonia. Mediastinal lymph node enlargement has been described in approximately 30 % of patients.
2.5 Differential Diagnosis
The differential diagnosis with other disorders manifesting with acute/subacute diffuse GGO e.g., opportunistic infections, pulmonary edema, and cellular nonspecific interstitial pneumonia (NSIP) may be difficult. However in HP the frequent association of lobular air trapping and centrilobular snowflake nodules is crucial for the diagnosis.
The so-called head-cheese pattern may also be observed in respiratory bronchiolitis-associated ILD (RB-ILD). Integrating nicotine poisoning and laboratory findings may indicate the most likely diagnosis (Chong et al. 2014).
GGO combined with cysts resemble those seen in lymphocytic interstitial pneumonia (LIP). However, LIP is often associated with other conditions, such as connective tissue diseases or lymphatic disorders (e.g., human immunodeficiency virus infection, lymphoma) (Ichikawa et al. 1994).
2.6 Management and Treatment
Patients with acute/subacute HP, if correctly and timely diagnosed and treated, generally have an excellent prognosis. The most important recommended therapy is inhibiting exposure to the causal agent by eliminating it from the environment, avoiding settings where it is present, or using a respirator in those settings. Systemic corticosteroids for a few days to weeks may improve symptoms. Indications for the use of such drugs include acute, severe, or progressive disease (Kokkarinen et al. 1992).
3 ARDS
3.1 Introduction
Acute respiratory distress syndrome (ARDS) is a condition characterized by sudden onset of severe hypoxemia and diffuse pulmonary infiltrates.
The syndrome was firstly introduced by Ashbaugh and colleagues in 1967 (Ashbaugh et al. 1967), while the criteria for the diagnosis of ARDS were first established in 1994 by the American-European Consensus Conference (AECC) (Bernard et al. 1994).
In 2012 the “Berlin definition,” the new updated consensus definition of ARDS, has been published in a high-impact journal. ARDS is defined as: “[…] type of acute diffuse, inflammatory lung injury, leading to increased pulmonary vascular permeability, increased lung weight, and loss of aerated lung tissue. The clinical hallmarks are hypoxemia and bilateral radiographic opacities […]” (ARDS Definition Task Force 2012).
3.2 Mechanisms of Injury
ARDS may follow several different types of lung injury that ultimately determine the same monomorphic pulmonary response, characterized histopathologically by the presence of diffuse alveolar damage (DAD).
There are many triggering events that have been classically classified as pulmonary or extrapulmonary. Pulmonary or direct injuries are processes determining direct injury to lungs, like infection, gastric aspiration, or toxic inhalation. Extrapulmonary or indirect injuries may be systemic processes like polytrauma, drug toxicity, sepsis, transfusions, or extra-thoracic diseases like acute pancreatitis.
From a histopathologic point of view, “diffuse” refers to the involvement of the whole alveolar structure: endothelium, basal membrane, and epithelium. The process is widespread throughout the lungs, but not always in a homogeneous manner: frequently there is presence of spared areas (Kligerman et al. 2013).
The process is characterized by a sequence of phases. Not necessarily the process develops through all the phases: it can stop and reverse anytime (Castro 2006).
The early or exudative phase lasts up to 7 days and is characterized by the presence of hyaline membranes. The alveolar epithelium is damaged and the basal membrane is exposed.
The next stage is the organizing, or proliferative, phase, characterized by the presence of organizing tissue and fibrosis. If this phase does not resolve favorably, there is progression to the last phase, the fibrotic phase.
The pattern of fibrosis is atypical and usually with predilection of the anterior segments (anti-gravitational distribution). Some patients may show areas of “honeycombing,” more frequently encountered following acute interstitial pneumonia (AIP), which is the “idiopathic” form ARDS (Tomiyama et al. 2001).
3.3 Terminology and Clinical Issues
The term ARDS should not be confused with the term permeability edema and it is not interchangeable with the term noncardiogenic edema. ARDS is the most severe form of permeability edema, but there are other forms of permeability edema without DAD (Ketai and Godwin 1998). Classification of pulmonary edema is discussed in the “Acute pulmonary edema” section.
ARDS, which is a life-threatening condition, is associated histopathologically to DAD.
In the AECC definition, ARDS was defined as an acute onset of hypoxemia, without specifying a timeframe to define acute; the PaO2/FiO2 (ratio of partial pressure of arterial oxygen to fraction of inspired oxygen) must be under <200 mmHg, with presence of bilateral infiltrates on frontal chest X-ray. Presence of left atrial hypertension must be ruled out. PaO2/FiO2 between 200 and 300 is termed acute lung injury (ALI).
When idiopathic, the process is termed acute interstitial pneumonia (AIP), also known as Hamman-Rich syndrome (Kliegerman et al. 2013). AIP may sometimes present with a more subacute course, and as a result, it does not always fulfill the criteria of ARDS (Janz et al. 2000).
The new definition of Berlin introduces several important changes to the old criteria (ARDS Definition Task Force 2012). The timeframe for define acute onset is specified within a week from a determinate event. A minimum level of positive end-expiratory pressure (PEEP) is established to evaluate the severity of the respiratory failure. The need to measure pulmonary wedge pressure is removed and the term ALI is suppressed. ARDS is now classified in three grades: mild (200< PaO2/FiO2 <300 with PEEP or C-PAP >5 cm H2O), moderate (100< PaO2/FiO2 <200 with PEEP >5 cm H2O), and severe (PaO2/FiO2 <100 with PEEP >5 cmH2O). Finally, it is stated that the pathologic correlate of ARDS is DAD.
A set of training radiograph is attached to improve interobserver reliability (Ferguson et al. 2012). Opacities seen on CT scan may substitute chest X-ray evaluation for the diagnosis of diffuse pulmonary infiltrates (Table 3).
AECC 1994 | Berlin 2012 | |
|---|---|---|
Onset | “Acute” (no specific timeframe) | Acute “within 7 days of a known clinical insult” |
Radiological criteria | Bilateral infiltrates on frontal CXR | “Bilateral infiltrates on frontal CXR not fully explained by effusions, lobar/lung collapse, or nodules /masses” (chest X-ray interpretation set available online) |
CT | Not included | Bilateral opacities on CT |
Pulmonary artery wedge pressure (PAWP) | PAWP <18 mmHg | Removed |
Severity | Acute lung injury (ALI) if PaO2/FiO2 <300 (regardless of PEEP level) ARDS PaO2/FiO2 < 200 (regardless of PEEP level) | ALI removed Mild/moderate/severe ARDS Minimum C-PAP/PEEP level 5 cmH2O |
Pathologic correlate | Injury to both the epithelium and the endothelium | DAD (diffuse alveolar damage) |
3.4 Imaging
3.4.1 Radiological Signs
The chest X-ray picture of ARDS varies depending on the phase of the process (Sheard et al. 2012).
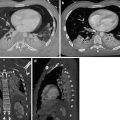
In the early or exudative phase, there is some degree of clinical-radiological dissociation, with a latent period after the injury of about 24 h, when the radiograph results normal (Fig. 5a).
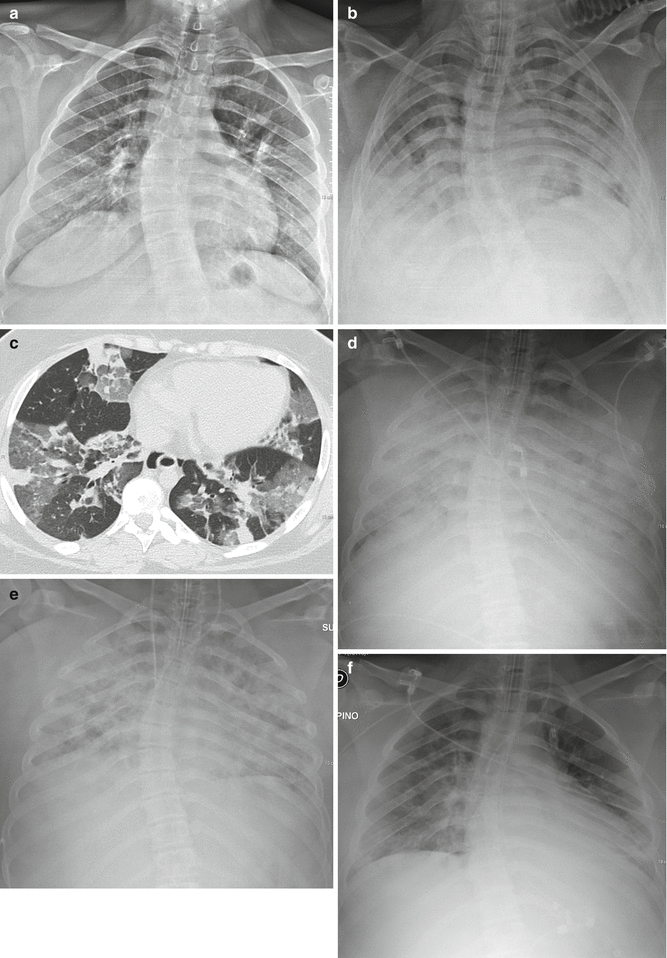
Fig. 5
Evolution of ARDS at chest X-ray (post-infective ARDS). Latent period: no pathologic findings (a). The following day appearance of bilateral patchy consolidations, with bilateral pleural effusion (b). HRCT, performed on the same day, shows bilateral patchy and lobular GGOs and consolidations, randomly distributed (atypical ARDS) (c). The day after, chest X-ray shows bilateral extensive opacification (“lung white-out”) (d). Parenchymal opacification remains stable: chest X-ray 2 days later (e). The patient survived and improved quickly: marked radiographic improvement 7 days later (f)
Subsequently the appearance of radiographic changes is rapid. The opacification is diffuse and symmetric, peripheral, and with presence of air-bronchograms. The heart and the vascular pedicle are not enlarged; pleural effusion is absent or limited (Fig. 5b). Septal thickening and peribronchial cuffing are less common than in HPE.
The picture remains stable for some days, or longer, during the proliferative phase. In more severe cases, the opacification is complete and the picture is that of the so-called “white lungs” (Fig. 5d, e).
In the late phase the alterations begin to reverse. In patients who heal, the lungs return normal (Fig. 5f), while in patients who develop fibrosis, there is evidence of coarse reticular opacities.
3.4.2 HRCT Signs
The HRCT pattern of ARDS depends on the phase of the process, even if we cannot clearly distinguish the phases. In particular, the exudative phase and the early organizing phase of ARDS overlap, as well as the late proliferative phase overlap with the fibrotic phase (Ichikado 2014).
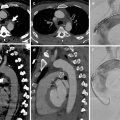
In the very beginning (early exudative phase), HRCT shows patchy ground-glass opacities (GGO) and consolidations, with geographic distribution (Figs. 5c and 6b). Small pleural effusions (missed by chest X-ray), septal pattern, and crazy paving can also be seen.
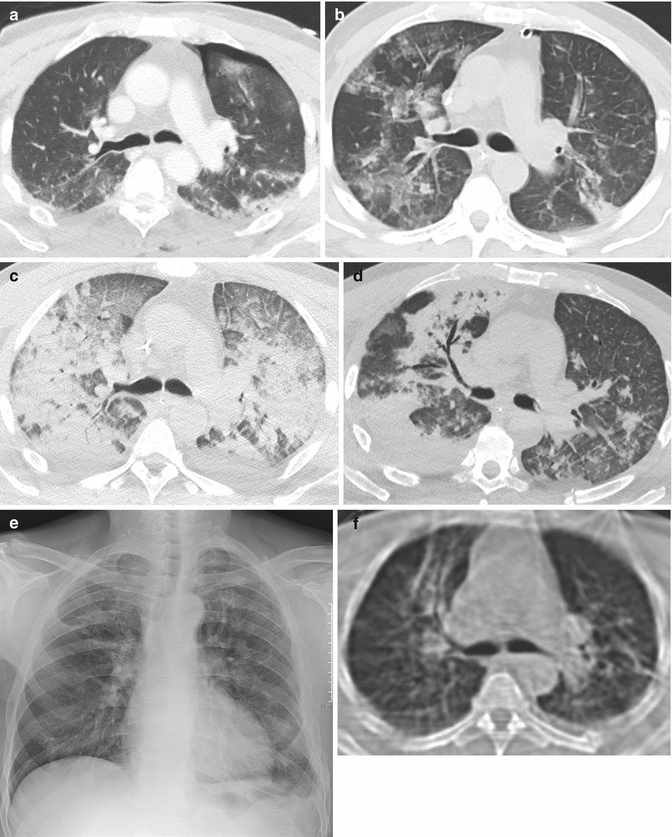
Fig. 6
Evolution of ARDS at CT (posttraumatic ARDS). At admission, left thoracic trauma with pneumothorax and lung contusion (a). Respiratory deterioration 7 days later: at CT appearance of bilateral patchy GGO, corresponding to the early exudative phase (b). Two days later, extensive lung opacification with extensive dorsal consolidation and ventral GGO: typical ARDS pattern (proliferative or organizative phase). Note presence of smooth septal thickening (“crazy-paving appearance”) (c). Nine days later advanced regression of the lung opacities: on the right side presence of bronchiectasis and bronchocentric consolidation, located anteriorly (fibro-proliferative phase) (d). The patient survived and recovered well, although chest X-ray performed 2 months later demonstrates persistence of coarse fibrosis in the upper right lobe (e), confirmed at CT (f)
Afterward, the process quickly progress to a more homogeneous opacification of the lungs, which persists for the exudative phase and the organizing phase. At this stage ARDS may show two patterns, named typical and atypical. Originally the typical pattern has been associated with extrapulmonary ARDS, while the atypical was associated with pulmonary ARDS (Goodman et al. 1999). This has not been confirmed by a following study (Desai et al. 2001), and in any case, it is not always possible to attribute a single cause to the insurgence of this syndrome (Desai 2002).
The HRCT pattern of typical ARDS is characterized by the presence of a bilateral symmetric anterior-posterior density gradient (Fig. 6c). The density is lower anteriorly (or may be normal or hyperventilated), while posteriorly the density increases progressively, from a ground-glass opacification to a frank consolidation, in the more dependent regions.
In atypical ARDS there are patchy GGO or consolidations, randomly distributed in dependent and nondependent regions of the lungs, without the symmetric density gradient (Fig. 5c).
In the late proliferative and fibrotic phase, from 38 to 85 % of the patients show fibrotic changes in the parenchyma, characterized by an atypical coarse reticular pattern, with traction bronchiectasis and bronchiolectasis (Fig. 7). Fibrotic alterations are more frequently distributed in the ventral portions of the lungs (Fig. 6e, f). This is considered to be the consequence of the barotrauma caused by prolonged ventilation (Nöbauer-Huhman et al. 2001).
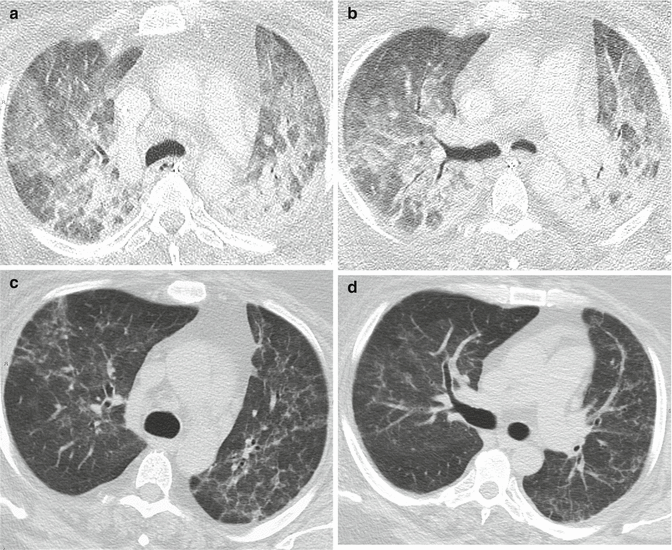
Fig. 7
Severe ARDS followed by fibrotic outcome, caused by viral H1N1 infection. Extensive bilateral consolidations and GGO (a, b). Two months later, persistence of a reticular pattern bilaterally (“atypical” fibrosis) (c, d)
CT predictors of mortality in ARDS are signs of right heart failure, involvement of more than 80 % of the lung parenchyma, and presence of varicoid traction bronchiectasis (Chung et al. 2011).
3.5 Differential Diagnosis
Historically, the radiographic differentiation of hydrostatic and permeability edema has always been a big radiological challenge (Milne et al. 1985; Aberle et al. 1988).
Criteria for the radiographic differentiation of permeability edema from hydrostatic pulmonary edema (HPE) have been discussed in the acute pulmonary edema paragraph. Although there are several signs that can help the diagnosis, the distinction is frequently impossible. Moreover, chest radiograph interpretation shows poor interobserver reliability (Rubenfeld et al. 1999; Meade et al. 2000).
Likely HRCT may improve the diagnostic accuracy, but at present there is a scarcity of literature in regard. HRCT findings that suggest presence of HPE are perihilar and upper lobar distribution of GGOs, central predominant distribution of consolidations, dilatation of the pulmonary veins and of the superior cava vein, thickening of the bronchial walls (peribronchovascular thickening), and right pleural effusion. Even distribution of the GGOs and of the consolidations is found more often in ARDS. Gravity-dependent opacities, septal pattern, air bronchograms, and traction bronchiectasis did not show significant difference in prevalence between HPE and ARDS (Komiya et al. 2013).
This difficulty in the differential diagnosis of the nature of the edema is implicitly recognized by the definition of Berlin. In the Berlin definition, a differential diagnosis between cardiogenic and noncardiogenic edema on chest radiograph is not required, nor it is considered relevant: it is now recognized that hydrostatic edema and ARDS may coexist. The diagnosis of ARDS requires that the respiratory failure is not fully explained by cardiac failure, or fluid overload, using all available data (Ferguson et al. 2012). Therefore, radiologically, the focus is on the generic diagnosis of bilateral edema that must not be confused with pleural effusions, lobar/lung collapse, or nodules/masses. In fact, HPE is not the only condition with which the ARDS goes in differential diagnosis: bioptic and autoptic studies have demonstrated only moderate agreement between the clinical diagnosis of ARDS and presence of DAD. Specificity of the various criteria for presence of DAD is variable in literature, but always quite poor (Lorente et al. 2015).
There is no pathognomonic laboratory test or radiological sign to establish the diagnosis of ARDS, which is generally clinical. Apart from HPE, the most common conditions that share a similar clinical presentation are atelectasis, pneumonia, and pulmonary embolism (Murray 1975). In reality there are many more and encountered less frequently: miliary tuberculosis, CMV pneumonia, invasive aspergillosis, hantavirus pneumonia, herpes simplex pneumonia, bronchoalveolar cell carcinoma, drug toxicity, lymphangitis, acute leukemia, lymphoma, veno-occlusive disease, sickle lung, acute eosinophilic pneumonia, acute cryptogenic organizing pneumonia (COP), acute fibrinous organizing pneumonia (AFOP), diffuse alveolar hemorrhage, and acute hypersensitivity pneumonia (Dakin and Griffiths 2002; Schwarz and Albert 2004).
Some of the abovementioned conditions may show peculiar HRCT findings (many are discussed elsewhere in this book). For example, presence of subpleural peripheral distribution of the consolidations is suggestive of acute eosinophilic pneumonia, while peribronchial distribution is suggestive of COP or AFOP (Obadina et al. 2013).
Most of those mimickers (or imitators) of ARDS require a different and specific therapy. Therefore, when performing a HRCT examination in patients with clinical diagnosis of ARDS, maximum attention is required to highlight any sign, or pattern, that may suggest an alternative diagnosis to the complex ARDS/DAD.
3.6 Management and Treatment
No pharmacologic therapy has proved effective in the prevention or management of ARDS.
The only specific therapy for ARDS is ventilation (noninvasive or mechanical) using low tidal volumes (lung-protective strategy), to improve blood oxygen levels, and providing supportive care.
Great deal of attention must be placed to not miss a treatable cause of ARDS and to early diagnose the complications (barotrauma, ventilator-associated pneumonia, and fluid overload, among others).
4 Diffuse Alveolar Hemorrhage (DAH)
4.1 Introduction
Diffuse alveolar hemorrhage (DAH) is not a specific disorder, but a syndrome that suggests a differential diagnosis and a specific sequence of testing. DAH may be a life-threatening condition characterized clinically by the presence of hemoptysis, falling hematocrit, diffuse pulmonary infiltrates, and hypoxemic respiratory failure, which can be severe. However, chest radiographic and CT findings may be nonspecific (the alveolar infiltrates can even sometimes be unilateral), and hemoptysis may be lacking. DAH should be considered a medical emergency due to the morbidity and mortality associated with failure to treat the disorder promptly (Collard and Schwarz 2004; Lara and Schwarz 2010).
The diagnosis of diffuse alveolar hemorrhage is made on the basis of the clinical and radiological pattern and may be confirmed by bronchoalveolar lavage (BAL). In acute and severe forms, the BAL findings include bright red blood from multiple sites in different bronchi and, microscopically, hemosiderin-laden macrophages. BAL is also useful for excluding other differential diagnoses, such as infections or other endobronchial sources of bleeding (Park 2013). Once the diagnosis is established, the underlying cause must be established in order to initiate treatment.
4.2 Mechanisms of Injury and Causes
DAH is a life-threatening condition which refers to hemorrhage originating in the pulmonary microvasculature, rather than from the bronchial circulation or parenchymal abnormalities. All causes of DAH have the common denominator of an injury to the alveolar microcirculation. Pulmonary small vessel vasculitides, connective tissue disorders, and drugs make up the majority of the cases of DAH (Table 4).
Table 4
Causes of diffuse alveolar hemorrhage (DAH)
Main causes of diffuse alveolar hemorrhage |
Pulmonary small vessel vasculitides: |
ANCA-associated pulmonary vasculitides: |
Granulomatosis with polyangiitis (formerly Wegener’s granulomatosis) |
Microscopic polyangiitis (MPA) |
Churg-Strauss vasculitis |
Non-ANCA-associated pulmonary vasculitides: |
Goodpasture’s syndrome |
Connective tissue disorders (SLE, mixed connective tissue disease, RA) |
Drugs |
Other causes of diffuse alveolar hemorrhage |
Coagulative disorders |
Inhaled toxins |
Mitral valve disease |
Pulmonary veno-occlusive disease (PVOD) |
Infection |
Diffuse alveolar damage |
Malignancy |
Autologous bone marrow transplantation |
Acute lung transplant rejection |
Pulmonary hemosiderosis |
Antiphospholipid syndrome |
DAH includes diseases associated with pulmonary capillaritis (cellular infiltrate of neutrophils in the capillaries and venules) and those associated with normal vessels (Colby et al. 2001). A pulmonary capillaritis is considered the most common underlying lesion associated with diffuse alveolar hemorrhage.
4.3 Terminology and Clinical Issues
The cardinal sign of DAH, hemoptysis, may be a dramatic event or evolve over days to weeks; however, it may be initially absent in up to 33 % of DAH cases. The other symptoms of DAH are nonspecific and include fever, cough, and dyspnea. Nonpulmonary signs and symptoms are those that accompany the underlying systemic disease. A possible association of hematuria and renal failure due to concomitant glomerulonephritis may be present (Dalpiaz et al. 2003).
The causes of diffuse alveolar hemorrhage are many, but the association with primary renal involvement (pulmonary-renal syndrome) reduces the range of diagnostic possibilities to a handful of diseases: Goodpasture’s syndrome, systemic lupus erythematosus (SLE), ANCA-associated vasculitis (antineutrophil cytoplasmic antibodies), particularly micropolyangiitis (MPA), granulomatosis with polyangiitis (Wegener granulomatosis), and, more rarely, eosinophilic granulomatosis with polyangiitis (Churg-Strauss) (Jennette 2013; Chung et al. 2010; Marten et al. 2005; Mayberry et al. 2000). Knowledge of the patient’s clinical data and laboratory tests may be sufficient to guide toward the possible cause of this group of patients (Table 5).
Table 5
Causes of combination of DAH and glomerulonephritis (primary pulmonary-renal syndrome)
Clinical features | Laboratory tests | |
|---|---|---|
Goodpasture’s syndrome | Young man | Anti-GBM |
Granulomatosis with polyangiitis (Wegener granulomatosis)
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|