This article focuses on pathologic processes affecting the optic nerve, optic nerve/sheath complex, and chiasm. Pathology of the visual pathways posterior to the chiasm is beyond the scope of this article and is discussed only briefly.
Clinical signs and symptoms
Decrease in visual acuity and visual field defects are the cardinal symptoms of optic nerve lesions. Their mode of onset depends on the underlying pathology: slowly growing compressive and infiltrative lesions lead to progressive visual loss, whereas ischemia, demyelinating lesions, trauma, and a sudden increase in orbital pressure can lead to rapid visual loss. The type of field defect depends on the location of the lesion. Lesions of the intraorbital portion of the optic nerves usually start with a central scotoma that may spread peripherally. Compression of the optic nerve at the level of the orbital apex may lead to abnormalities in a localized sector. Loss of color vision may be an early sign in several optic nerve pathologies, including demyelination, tumors, and toxic neuropathy. Intracranial compression of the optic chiasm classically leads to bitemporal hemianopia.
When examining patients who have suspected optic nerve lesions, it is important to have knowledge of visual field defects produced by postchiasmatic lesions. Selective involvement of the temporal fibers of the optic radiation (Meyer’s loop), which may occur in tumors or infarctions confined to the temporal lobe, results in a homonymous superior quandrantanopia. Posterior lesions in the optic radiation produce a hemianopia with macular sparing.
Involvement of the entire striate cortex of the occipital lobe results in a contralateral homonymous hemianopia. Lesions involving the inferior lip of the striate cortex present with a contralateral upper quadrantanopia, and those involving the superior lip present with a contralateral lower quadrantanopia.
Other symptoms associated with optic nerve pathology include proptosis (with intraorbital mass lesions), strabismus, oculomotor nerve palsies (which may be a feature of optic nerve gliomas or other orbital mass lesions and of raised intracranial pressure), headaches (idiopathic intracranial hypertension [IIH]), and conjunctival injection (arteriovenous fistulae). Pituitary lesions compressing the optic nerves and chiasm can be associated with endocrine symptoms.
Developmental and inherited conditions
Colobomas
Colobomas are a developmental abnormality arising from failure of closure of the fetal intraocular fissure during gestation. The defect can affect the iris, lens, ciliary body, retina, choroid, sclera, and optic nerve; optic nerve involvement may result in visual field scotomas.
Colobomas account for approximately 2% of congenital malformations of the eye and may occur as an isolated anomaly or part of a multisystemic syndrome. The posterior globe and optic nerve are involved most commonly and the cleft often is in the inferonasal quadrant. Up to 60% may be bilateral ( Fig. 1 A) and inheritance may be autosomal dominant, with variable penetrance.
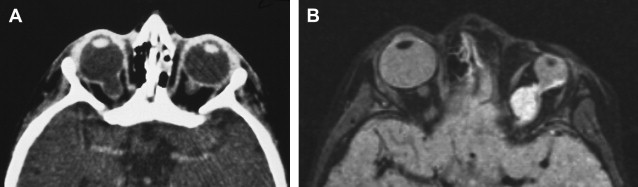
The shape of the globe and optic nerve coloboma depend on the extent of retinal inversion, and if there is extensive separation of the retinal layers in association with retinal proliferation, large cysts may form that have mass effect and are associated with micro-ophthalmos ( Fig. 1 B).
Systemic abnormalities and syndromes associated with colobomas include trisomy 13 and 18, Potter’s syndrome, CHARGE and VATER association, Joubert and Warburg syndromes, congenital heart disease, polydactyly, and holoprosencephaly. The renal-coloboma syndrome is associated with a mutation of the PAX2 gene.
Leber’s Hereditary Optic Neuropathy
Leber’s hereditary optic neuropathy, a hereditary condition resulting from degeneration of retinal ganglion cells and their axons, results in severe acute or subacute visual loss. It affects predominantly young men and is transmitted maternally. It is the result of several mutations in the mitochondrial genome with effects on the oxidative phosphorylation chain in mitochondria, but it remains unclear exactly how this causes death of cells in the optic nerve head.
Patients usually present with acute unilateral visual loss, followed by a similar event in the contralateral eye several weeks later, and the end result is severe optic atrophy and a permanent reduction in visual acuity. More than 50% of men and 85% of women who have the genetic mutations do not experience visual loss, however.
MRI of the anterior visual pathway in the acute stages may be normal, but in later stages, optic atrophy and increased T2 signal in the optic nerves are present. Swelling and enhancement of the nerves is reported in the acute stage, and demyelinating/multiple sclerosis–like lesions are also encountered on cranial MRI.
Other inherited optic neuropathies, such as dominant optic neuropathy, hereditary spastic paraparesis, and Friedreich’s ataxia share the mechanism of mitochondrial dysfunction.
Developmental and inherited conditions
Colobomas
Colobomas are a developmental abnormality arising from failure of closure of the fetal intraocular fissure during gestation. The defect can affect the iris, lens, ciliary body, retina, choroid, sclera, and optic nerve; optic nerve involvement may result in visual field scotomas.
Colobomas account for approximately 2% of congenital malformations of the eye and may occur as an isolated anomaly or part of a multisystemic syndrome. The posterior globe and optic nerve are involved most commonly and the cleft often is in the inferonasal quadrant. Up to 60% may be bilateral ( Fig. 1 A) and inheritance may be autosomal dominant, with variable penetrance.
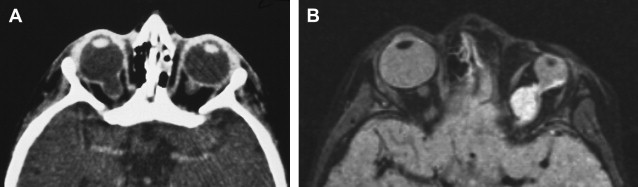
The shape of the globe and optic nerve coloboma depend on the extent of retinal inversion, and if there is extensive separation of the retinal layers in association with retinal proliferation, large cysts may form that have mass effect and are associated with micro-ophthalmos ( Fig. 1 B).
Systemic abnormalities and syndromes associated with colobomas include trisomy 13 and 18, Potter’s syndrome, CHARGE and VATER association, Joubert and Warburg syndromes, congenital heart disease, polydactyly, and holoprosencephaly. The renal-coloboma syndrome is associated with a mutation of the PAX2 gene.
Leber’s Hereditary Optic Neuropathy
Leber’s hereditary optic neuropathy, a hereditary condition resulting from degeneration of retinal ganglion cells and their axons, results in severe acute or subacute visual loss. It affects predominantly young men and is transmitted maternally. It is the result of several mutations in the mitochondrial genome with effects on the oxidative phosphorylation chain in mitochondria, but it remains unclear exactly how this causes death of cells in the optic nerve head.
Patients usually present with acute unilateral visual loss, followed by a similar event in the contralateral eye several weeks later, and the end result is severe optic atrophy and a permanent reduction in visual acuity. More than 50% of men and 85% of women who have the genetic mutations do not experience visual loss, however.
MRI of the anterior visual pathway in the acute stages may be normal, but in later stages, optic atrophy and increased T2 signal in the optic nerves are present. Swelling and enhancement of the nerves is reported in the acute stage, and demyelinating/multiple sclerosis–like lesions are also encountered on cranial MRI.
Other inherited optic neuropathies, such as dominant optic neuropathy, hereditary spastic paraparesis, and Friedreich’s ataxia share the mechanism of mitochondrial dysfunction.
Trauma
Trauma of the anterior optic pathways includes direct and indirect injury to the optic nerve. Direct injury is caused most frequently by objects penetrating the orbit ( Fig. 2 ) or by bone fragments at the orbital apex. Deceleration injury to the optic nerves and chiasm can be a feature of severe head trauma. The traumatic chiasmal syndrome frequently results in blindness in one eye and a temporal defect in the other or in bitemporal field defects. Soft tissue injuries and bleeding can lead to a rapid rise in intraorbital pressure with secondary (indirect) compressive and ischemic injury to the optic nerve. Shearing forces also may lead to hemorrhage into the optic sheath, causing profound visual symptoms with minimal orbital signs.

Traumatic injury of the globe and optic nerve is assessed best with CT, including multiplanar reformatted images. CT is able to demonstrate intraocular and intraorbital hemorrhage, foreign bodies, and bony fragments. MRI should be avoided if the injury involves metallic fragments, which may be deflected in a magnetic field, thereby causing further damage.
Trauma to the optic nerve also may result from thermal, chemical, or radiation injury. Damage to the optic nerve can occur after radiotherapy of sellar and parasellar tumors and of lymphoproliferative and leukemic disease of the orbit. Radiation-induced optic neuropathy can involve any part of the optic nerve, including the optic nerve head ( Fig. 3 A), and, not infrequently, the intracranial portion of the optic nerve ( Fig. 3 B). The affected portions are T2 hyperintense and may show contrast enhancement.
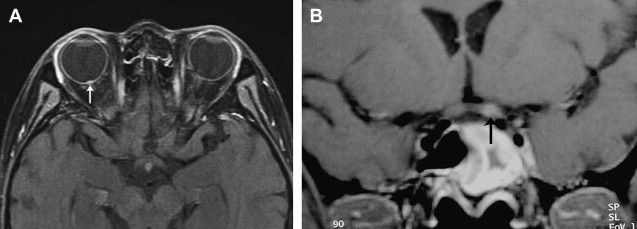
Vascular lesions
Ischemia
The retina and optic nerve head are sensitive to ischemia, which may lead to transient monocular blindness (amaurosis fugax) or to a more permanent visual deficit. In young patients, consider ophthalmic migraine, but in older patients, ischemic ocular symptoms usually are caused by a stenosis or occlusion of the internal carotid artery and retinal artery emboli. Retinal vein occlusion also may cause ischemic symptoms, predisposing conditions being lupus erythematosis or phospholipid syndrome.
Caroticocavernous Fistulae
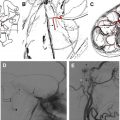
Caroticocavernous fistulae (CCF) cause raised pressure and enlargement of the ophthalmic veins ( Fig. 4 A, B), leading to proptosis and raised intraocular pressure. If sufficiently high, the raised intraocular pressure interferes with the vascular supply and function of the optic nerve and can lead to monocular blindness if not treated in time. Direct CCF are the result of a tear in the wall of the cavernous portion of the internal carotid artery, which creates a direct communication with the cavernous sinus, and are post-traumatic in the majority of cases. Indirect CCF are the result of dural arteriovenous fistulae in the wall of the cavernous sinus, usually supplied by multiple feeders from the external or internal carotid arteries. Indirect CCF may develop spontaneously or secondary to cavernous sinus thrombosis. Direct and indirect CCF reverse the flow in the ophthalmic veins and increase the intraocular pressure. Both types of CCF can be treated by endovascular means. Direct CCF are treated most commonly with occlusion of the tear by balloons inserted from the arterial side, and indirect dural fistulae are treated most commonly with coil embolization of the cavernous sinus from the venous side.
Aneurysms
Aneurysms arising from the cavernous and supraclinoid segments of the internal carotid artery can present with visual symptoms resulting from their mass effect on the optic nerves and optic chiasm ( Fig. 5 ). Giant aneurysms in this region are delineated best with contrast-enhanced magnetic resonance angiography or CT angiography. Endovascular treatment options include occlusion of the parent artery (after a successful balloon test occlusion) and coiling. The former leads to a more immediate decrease in mass effect; the latter reduces the pulsatility of the mass in a first instance, which is associated with clinical improvement.
Wyburn-Mason Syndrome
Wyburn-Mason syndrome is a rare congenital neurocutaneous disorder with facial nevi and ipsilateral arteriovenous malformations (AVMs), which can occur anywhere along the visual pathways from the occipital cortex to the orbit. A recent review of 27 cases found orbital AVMs in 17 cases and involvement of the optic chiasm in seven. Decrease in visual acuity is not uncommon in Wyburn-Mason syndrome, with optic nerve compromise caused by compression by an expanding AVM or ischemia.
Tumors of the optic nerve
Primary Tumors of the Optic Nerve
The most frequent primary tumors of the optic nerve are gliomas and optic nerve sheath meningiomas, the former four times more common. Other primary optic nerve tumors are rare and include hemangioblastomas, hemangiopericytomas, medulloepitheliomas, and gangliogliomas.
Optic Nerve Gliomas
Optic nerve gliomas originate from astrocytes of the optic nerve and the majority of these tumors occur below age 20 with a peak incidence of from 2 to 8 years. In children, these tumors have a benign histology (pilocytic astrocytomas) and slow growth rate. The more rare adult subgroup has a malignant histology with features of an anaplastic astrocytoma or glioblastoma mutliforme and rapidly may invade adjacent structures, such as the hypothalamus or temporal lobes.
Gliomas can occur anywhere along the optic pathway and present with loss of visual acuity or color vision, abnormal pupillary function, or painless proptosis, depending on their location.
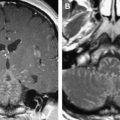
Fusiform enlargement, kinking, and tortuosity of the optic nerve and enlargement of the optic canal are typical features on CT and MRI. Unlike optic nerve sheath meningiomas, gliomas do not show calcification on CT. On MRI, they appear hypo- or isointense on T1- and hyperintense on T2-weighted images and enhance to a variable degree ( Fig. 6 A, B).
Optic nerve gliomas also are a feature of neurofibromatosis 1 (NF1) and bilateral optic nerve gliomas virtually are diagnostic of this condition ( Fig. 7 A–C). Patients who have NF1 may show other intracranial lesions, such as hyperintensities in the basal ganglia, brainstem, and cerebellum or astrocytomas. There is increasing evidence that optic pathway gliomas behave differently in patients who have and who do not have NF1. In NF1 patients optic gliomas involve most frequently the optic nerve, whereas in patients who do not have NF1, the optic chiasm represents the most common site ( Fig. 8 A, B). In patients who do not have NF1, the tumors are less stable on follow-up imaging and more likely to extend outside the optic pathways. Visual loss is more common in patients who do not have NF1 (80%) compared with those who have NF1 (20%). There are many well-documented cases of spontaneous regression of optic nerve gliomas in NF1.