Chapter 50 With the advances in high-resolution ultrasonography and magnetic resonance imaging (MRI), thoracic lesions are now commonly detected before birth. The most common congenital thoracic lesions include congenital diaphragmatic hernia (CDH); congenital bronchopulmonary malformations, which represent a group of lung anomalies, including congenital pulmonary airway malformation (CPAM), bronchopulmonary sequestration (BPS), and congenital lobar overinflation (CLO); and congenital hydrothorax. Additionally, congenital high airway obstruction (CHAOS), which is caused by obstruction or absence of the trachea, may also present in utero. Pulmonary hypoplasia, agenesis, and aplasia are less common and discussed in Chapter 53.1–4 In a fetus, the clinical importance of these lesions lies primarily in the mass effect on surrounding structures. This may result in compression of the airway, blood vessels, lymphatics, and lung with development of pleural effusions, polyhydramnios, hydrops, and pulmonary hypoplasia. The outcome depends on the timing of secondary effects and the severity of pulmonary hypoplasia. With improvements in fetal imaging, fetal intervention has also advanced. Masses that resulted in hydrops in the past were typically lethal. Now these masses may be amenable to fetal intervention including maternal steroids, intrauterine cyst aspiration, fetal thoracentesis, thoracoamniotic shunts, laser therapy, sclerotherapy, and in utero fetoscopic or open fetal surgery.5–9 In maternal-fetal surgery, the greatest responsibility is to the mother, who is an innocent bystander in the maldevelopment occurring in utero. She must be aware of the risks and benefits for herself and her infant and has the potential to undergo an operation that has no direct benefit to her. The limiting factor of invasive fetal therapy continues to be preterm labor despite advances in uterine relaxation. Almost all fetuses that undergo fetal therapy are still delivered prematurely. Women who have undergone open fetal surgery have been able to have normal children subsequently, although delivery of subsequent pregnancies requires cesarean section. Thus, invasive fetal therapy is limited to circumstances in which it may be predicted with reasonable certainty that the fetus or infant would not survive without fetal intervention.7 Congenital Diaphragmatic Hernia Etiology: The diaphragm develops in the fourth week of gestation with fusion of four components. By 8 weeks, the pleural–peritoneal cavity separates. There is caudal migration with final diaphragmatic positioning by 12 weeks. The most common CDH occurs secondary to failure of fusion of the left pleuroperitoneal membrane to the remaining primitive diaphragm (Bochdalek hernia). The bowel returns to the abdomen by the 10th week of gestation and may begin to herniate into the chest if a diaphragmatic defect is present. CDH occurs in 1 : 3000 to 1 : 4000 live births. They occur on the left in 88%, the right in 10%, and bilaterally in 2%. Herniation occurs by the second trimester and affects early lung development on the contralateral and ipsilateral sides. CDH is typically sporadic. However, a 15% to 45% incidence of associated anomalies is seen. Rare familial cases and syndromes have been described.10 The presence of a major malformation significantly increases the mortality rate. Thus, close assessment of additional anomalies, fetal echocardiography, and karyotyping are important for appropriate counseling. Imaging: The diagnosis of CDH in fetuses may be missed ultrasonographically, particularly when the stomach is not herniated into the chest.11,12 In these cases, mediastinal shift may be the first finding to suggest the diagnosis. Ultrasonographic evaluation should characterize the location of the diaphragmatic defect and the amount of herniated organs. Coronal and sagittal images help locate the defect, whereas axial images are important to assess the amount of mediastinal deviation. Contralateral lung volume should be measured. Ipsilateral lung tissue should be looked for but typically is too compressed to be visualized. The stomach and gallbladder are identified, and the location above or below the diaphragm is noted. Herniation of the liver into the chest is associated with a poorer outcome and thus is important to determine. This may be difficult ultrasonographically because the liver and lung may have a similar echotexture. An abnormal course of the umbilical, hepatic, and portal veins may help confirm this finding.13 With left CDH, the left lobe of the liver typically herniates into the anterior chest. If the stomach is adjacent to the anterior chest wall, liver herniation is unlikely. With right-sided hernias, the liver is more posterior in the chest. At times, small bowel loops may mimic lung cysts, making it difficult to differentiate it from CPAM. Color Doppler of the superior mesenteric artery may be helpful in confirming herniation of bowel loops by following the vessel into the chest. Associated anomalies, particularly cardiac, should be searched for. Fetal MRI protocol should include T2-weighted images in all three planes and T1-weighted breath hold gradient recalled echo images in the coronal plane to assess for liver and meconium positions (Fig. 50-1). Meconium-filled bowel loops are low signal on T2-weighted images and high signal on T1-weighted images, making location of the bowel easy. The liver is high signal on T1-weighted images and intermediate signal on T2-weighted images, also making it easy to separate from bowel and adjacent lung (e-Fig. 50-2). MRI is most helpful in the evaluation of right and bilateral CDH.14,15 In these cases, with the stomach often located below the diaphragm in the abdomen, it may be difficult to differentiate a CPAM from a CDH by ultrasound. MRI easily distinguishes abdominal contents within the chest from cystic lesions and provides specific information on hernia content, size of diaphragmatic defect, and amount of ipsilateral and contralateral lung. Figure 50-1 Left congenital diaphragmatic hernia. e-Figure 50-2 Congenital diaphragmatic hernia with covering membrane. One goal of fetal imaging is to predict outcome when counseling patients. Survival rates range from 40% to 90% with an overall mortality reported to be 70% to 80%.16 Currently, no single finding or measurement has been shown to be an absolute indicator of postnatal outcome. Indications for poor prognosis include large mediastinal shift and liver herniation.17 With liver herniation, the survival rate has been reported to be approximately 50%, whereas fetuses without herniation may have up to 90% survival.14,18,19 Additional findings such as intrathoracic location of the stomach, asymmetric size of cardiac ventricles, and polyhydramnios also may indicate a worse prognosis. Syndromes associated with CDH, for example, Fryns syndrome, have a poorer prognosis (e-Fig. 50-3).10 The presence of additional anomalies also affect outcome. Bilateral CDHs are typically fatal. e-Figure 50-3 Fryns syndrome. The degree of lung hypoplasia has prognostic value, but the best way to quantify this remains unclear. Measurement of the lung-to-head ratio adjusts for fetal size and gestational age and estimates the amount of residual contralateral lung.19–22 The cross-sectional area of the lung at the level of the four-chamber view of the heart is measured ultrasonographically and expressed as a ratio of lung cross-sectional area over head circumference. With a low lung-to-head ratio, the outcome is poor; with a high ratio, the outcome is more favorable. At the extremes, if the ratio is less than 0.6, mortality is 100%, and if the ratio is greater than 1.4, the survival rate is 90%. Multiple other methods have been attempted by MRI to quantify lung hypoplasia.23–27 Total lung volumes may be compared with expected lung volumes for age. A lung volume of greater than 25 cm3 suggests a favorable prognosis, whereas less than 18 cm3 suggests a poor outcome. A ratio of observed to predicted lung volume less than 25% suggests a poor outcome.26,27 Estimated expected lung volume is calculated by measuring fetal thoracic volume and then subtracting mediastinal volume. The lung volume of the fetus with a CDH is then measured and divided by this expected lung volume to give the percent predicted lung volume. Percent predicted lung volume of less than 15% has a 40% rate of survival compared to 100% if greater than 15%.28 Other studies have suggested, however, that calculated lung volume is not predictive of outcome.22,29,30 Infants with CDH may develop pulmonary hypertension, and this may be predicted by the modified McGoon index. From ultrasonographic or MRI measurements, the right and left pulmonary artery diameters are added, which is then divided by the aortic diameter. If the value is less than 0.8, the risk of developing pulmonary hypertension is high; if the value is greater than 1.0, the risk is low.31 Treatment and Follow-up: A wide spectrum of severity after birth exists in infants with the same anatomic defect, with some most likely to live and others most likely to die. The severity of pulmonary hypoplasia is determined by the timing of when the abdominal contents moved into the thorax and how much they compressed the developing lung. It is also known that esophageal obstruction from herniation and kinking of the gastroesophageal junction of the stomach produces polyhydramnios. To help these pregnancies, the fetal lung needs to be made to grow larger before birth, or a way has to be devised to support lung function after birth. Extracorporeal membrane oxygenation (ECMO) may support lung function for several weeks until the lungs grow and pulmonary hypertension resolves. To help lungs have more space to grow in utero, open fetal surgical diaphragmatic repair has been attempted. Most fetuses with CDH with their liver in the thorax are not amenable to traditional antenatal surgical repair because the umbilical vein kinks when the liver is returned to the abdomen. Open surgery worked fairly well for fetuses with livers “down” (below the diaphragm). For fetuses with livers “up” (above the diaphragm), however, this approach has failed. However, the National Institutes of Health (NIH) sponsored prospective fetal surgical trial showed no improvement in survival in fetuses with their livers “down.”32 The history of fetal intervention for CDH has evolved from surgically invasive procedures to less invasive fetoscopic techniques.29,33,34 It was noted that fetuses with laryngeal or tracheal atresia have larger than normal lungs. This led to animal experimentation that revealed that when the trachea was occluded and fetal lung fluid egress blocked, lung growth was stimulated. In utero tracheal occlusion, initially by application of an external tracheal clip and now by fetal bronchoscopic insertion of an occlusive balloon, has been performed to allow lungs to grow.35 At delivery, the EXIT (ex-utero intrapartum treatment) procedure is performed to remove the tracheal clip or balloon prior to separation from the placenta. The efficacy of this endoscopic approach has had mixed results. An NIH-sponsored randomized trial in the United States failed to show a significant advantage. Temporary tracheal occlusion via fetoscopy is now being attempted in some centers, with a suggestion of early favorable results. Currently, the standard of care includes supportive care after term delivery with high-frequency ventilation and ECMO, if needed, with some centers advocating an EXIT-to-ECMO approach for high-risk fetuses.36 Etiology: CPAM, previously known as congenital cystic adenomatoid malformation, is the most common congenital lung abnormality, representing 30% to 47% of lung masses diagnosed in utero. CPAM is a hamartomatous lesion characterized by abnormal growth of terminal bronchioles containing cystic and solid tissue. It is currently thought that maldevelopment of the airways results in obstructive dysplastic changes.37–39 Typically unilobular (85% to 95%), CPAMs may be multilobular and bilateral. They contain multiple microcysts or macrocysts lined by respiratory epithelium. Usually, a communication with the tracheobronchial tree exists, with blood supplied from pulmonary arteries. Stocker’s classification of CPAM includes five different types.40 This is based on cyst size and underlying histologic resemblance to the bronchial tree and airspaces. Adzick described a classification based on fetal imaging appearance and gross anatomy: (1) macrocystic CPAM (multiple large cysts >5 mm with slow growth and favorable prognosis); and (2) microcystic CPAM (cysts <5 mm with solid appearance which have the highest risk for developing hydrops).41 Imaging: Ultrasonographic appearance of CPAM depends on histology. With macrocystic CPAM, multiple nonconnecting cysts are seen. Often, a single lobe is affected, although multiple lobes and bilateral CPAM have been reported. Microcystic CPAMs are homogeneously echogenic and usually more echogenic than the adjacent lung.42 Doppler shows flow from PAs in contradistinction to BPS, in which the vascular supply is from the aorta. Hybrid lesions with mixed features of CPAM (macrocysts) and BPS (aortic supply) are common. If the echogenicity of the CPAM is similar to adjacent lung, mass affect may be the only finding to suggest the diagnosis.43 MRI is useful in delineating the lung mass and amount of residual normal lung. The lung malformation is typically of high signal with macrocysts and intermediate signal with microcysts (Fig. 50-4).44
Prenatal Imaging and Intervention
Common Congenital Thoracic Lesions in Fetus
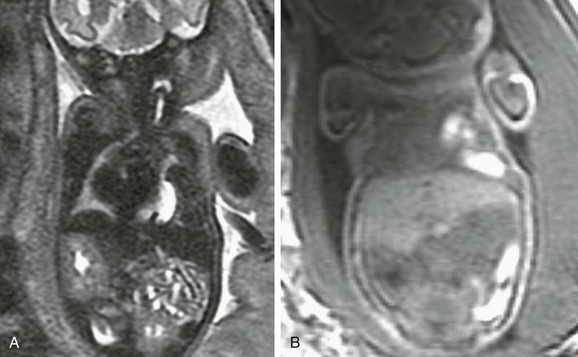
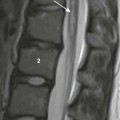
A, A coronal single-shot fast spin echo T2-weighted magnetic resonance image at 32 weeks’ gestation demonstrates deviation of the heart to the right. High-signal fluid-filled stomach and low-signal meconium are herniated into the left chest. The liver appears infradiaphragmatic. B, A coronal gradient recalled echo T1-weighted image shows high signal meconium-filled colon herniated into the left hemithorax. The intermediate signal liver is infradiaphragmatic.
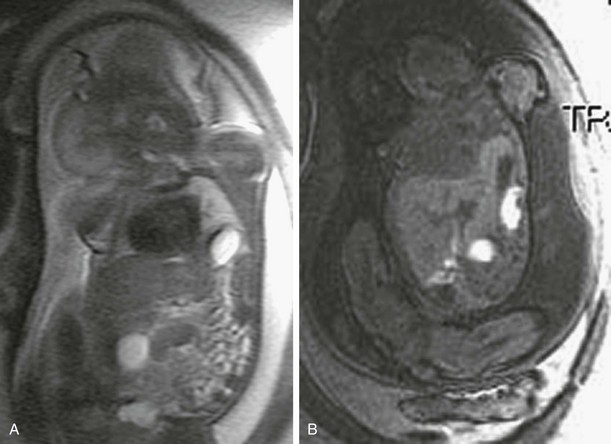
A, A coronal single-shot fast spin echo T2-weighted magnetic resonance image shows herniated stomach, bowel, and a portion of the left lobe of the liver herniated into the left hemithorax. Residual left lung parenchyma is noted superior to the stomach. B, A coronal gradient recalled echo T1-weighted magnetic resonance image confirms that high signal meconium-filled colon and intermediate high signal liver are herniated into the left hemithorax.
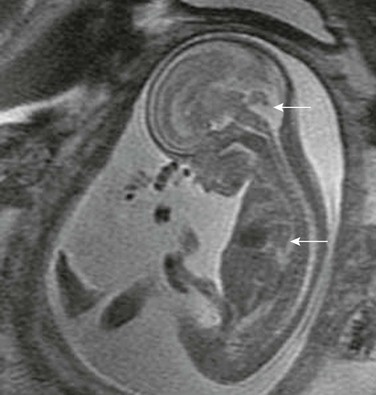
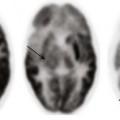
A single-shot fast spin echo T2-weighted right parasagittal magnetic resonance image of a fetus of 24-week gestational age shows bowel loops (inferior arrow) herniated into the hemithorax. Inferior vermian hypoplasia (superior arrow) and nuchal thickening are present. This infant was diagnosed with Fryns syndrome at delivery.
Congenital Pulmonary Airway Malformation
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree