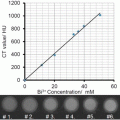
Fig. 1
Radioactive follow-up of nanoparticle core and shell in vivo . (a) The nanoparticles consist of 59Fe-labeled IONPs stabilized with 14C-labeled oleic acid and modified with DSPE-mPEG2000 and 111In-DMPE-DTPA phospholipid. (b) Comparison of the 59Fe, 111In, and 14C tissue concentrations (expressed as percentage of the injected radioactivity per gram) at different time points after intravenous injection of labeled IONPs. Adapted from Wang et al. [28] with permission
The biodistribution of 59Fe and 111In was broadly similar, with highest uptake in liver and spleen, especially at 24 h post-injection (Fig. 1). The greatest discrepancy was found in the kidneys, with an eightfold higher concentration of 111In compared to 59Fe. 111In was recovered at each time point in the urine suggesting the loss of 111In3+ ions or other 111In species. The 14C exhibited much lower concentration in liver and spleen, confirming a previous study by Freund et al. showing that oleic acid could detach from the iron oxide core in vivo in the bloodstream [25]. Three control experiments were necessary to assess the distribution of the different components of the shell: injection of the labeled phospholipid (DMPE-DTPA-111In), of the citrated label (111In-citrate), and of the labeled ligand (DTPA-111In). The phospholipid DMPE-DTPA-111In had a similar biodistribution as the 111In-labeled IONPs, but exhibited a lower accumulation in liver, spleen, and bone and a higher accumulation in the blood, heart , and lungs. In contrast, 111In citrate was found in the kidney and DTPA-111In in the urine. These results suggest that 111In is not released from NPs in 111In-citrate or DTPA-111In forms but rather as DMPE-DTPA-111In, which forms micelles with the same biodistribution as the nanoparticles . In agreement with this hypothesis, the 59Fe/111In ratio decreases from 2:1 at 10 min to 1.25 at 24 h in liver, confirming an incomplete and early dissociation of phospholipids from nanoparticles. In the work by Kreyling et al. the 198Au:111In ratio was also analyzed and revealed differences in biodistribution between the two isotopes, suggesting a partial dissociation of the coating from the nanoparticles [29]. The control 111In-DTPA was rapidly excreted in urine, while the ionic form of 111In accumulated in the liver. Most of the 111In and 198Au administered in particulate form are accumulated in the liver. However, the results could not indicate if the 111In detached from Au NPs was in the ionic form or still attached to the polymer [29]. A minor dissociation of the coating occurred in the bloodstream, but the liver could accumulate both intact NPs and separated components [29].
To go further in the analysis, in vitro tests have been performed to mimic the behavior of the NPs and their coating in the liver [29]. Polymer-coated Au NPs and CdSe QDots were accumulated in the lysosomes of Kupffer cells and endothelial cells. The co-localization of QDots and of the polymer shell labeled with DY495 was investigated by fluorescence, showing that the DY495 has been partly displaced from QDots and subsequently exocytosed after several hours [29]. This result suggested an intracellular enzymatic degradation of the polymer shell [30], but could not conclude whether there is a dissociation of the polymer from the nanoparticles or of the DY495 from the polymer or both.
Another relevant parameter to study the fate of the complex formed by nanoparticles and their coating is the presence of the protein corona , which evolves dynamically throughout the journey of the nanoparticle within the body. To understand the influence and the fate of the protein corona in vivo , Bargheer et al. used 59Fe-labeled PEGylated IONPs and 125I-labeled model proteins such as transferrin, covalently bound or adsorbed to the surface of the nanoparticles (Fig. 2) [26]. Importantly, both 125I and 59Fe labels were cleared synchronously from the blood and the biodistribution of 125I transferrin that was bound or adsorbed to IONPs was clearly different from the biodistribution of free 125I transferrin. This result suggests that the protein corona around the nanoparticles was not lost in the bloodstream in the first hour post-administration [26]. However, the ratio 125I/59Fe decreased in all organs after 2 h except in stomach and blood, suggesting a substantial recycling and transfer of the transferrin from the liver to the blood and other tissues. Interestingly, there was no significant difference between covalently bound and adsorbed transferrin in this experiment [26]. It must be noted that transferrin was among the adsorbed proteins, which show the highest abundance (>10 %) in coronae of many nanoparticle types [31].

Fig. 2
Radioactive follow-up of nanoparticle core and protein corona in vivo . 59Fe-labeled IONPs with a preformed corona of adsorbed (a, b, right side) or covalently bound (a, b, left side) 125I-mouse transferrin, i.v. injected to mice in comparison to free 125I-mouse transferrin. (a) Activity of 59Fe and 125I (1–120 min) in blood. (b, c) Activity of 59Fe and 125I in organs 120 min after injection of IONPs or free transferrin. Adapted from Bargheer et al. (open access) [26]
1.5 Cellular “Conditioning” of Administered Nanoparticles and Its Impact on Imaging Outcomes
By analogy with the extracellular environment (as for example the circulating blood plasma), which constantly supplies and exchanges proteins surrounding the nanoparticles, the cells continuously modify the conditions of their microenvironments and thus continuously reshape the protein corona of cell-internalized nanoparticles. Wang et al. studied the intracellular processing of fluorescently labeled protein corona at the cellular scale [32]. The adsorbed layer of proteins, resulting from incubation of positively charged polystyrene nanoparticles with full fluorescent serum, was strong enough to be retained on the nanoparticles as it entered the cells and was trafficked into the lysosomes together with nanoparticles (Fig. 3). Within the lysosomes , a progressive decrease of the protein corona fluorescence was observed, indicating the degradation of the protein corona in the lysosomes over 6–8 h after exposure. Interestingly, the nanoparticles were able to convey a much higher amount of proteins than a simple incubation of cells with proteins would provide [32]. This suggests that enzymatic digestion by proteases at the lysosomal level could be activated by nanoparticle uptake. Importantly, the clearance of protein fluorescence was accompanied by a lysosomal swelling and increased lysosomal permeability, with subsequent damages on cell organelles and initiation of apoptotic signaling pathways (Fig. 3) [32]. Thus the protein corona masks for a while and delays the deleterious effect of positively charged polystyrene nanoparticles until being degraded in the lysosomes by enzymes, such as the cathepsin L [29, 30].

Fig. 3
Intracellular fate of the protein corona . (a) Green fluorescent protein corona formed on the blue positively charged amino-modified polystyrene nanoparticles (PS-NH2 NPs, 50 nm) is retained after cellular uptake. (b) Time-resolved localization of PS-NH2 NPs and their impact on lysosomes in 1321N1 cells, showing increasing accumulation in the lysosomes and subsequent progressive swelling of lysosomes. (a PS-NH2 NPs (green), lysosomes (red: LAMP-1 antibody), nuclei (blue, DAPI staining); (b) PS-NH2 NPs (blue), lysosomes (green: LAMP-1 antibody), nuclei (red: Draq 5 staining)). (c) Kinetics of the degradation of the fluorescent-labeled protein corona of PS-NH2 NPs. (d) Flow cytometry distribution of LysoTracker Red staining after exposure to PS-NH2 NPs, indicating increasing volume of lysosomal volume and gradual formation of a second population with loss of lysosomal integrity. (e) Western blot of different cathepsins in cytosolic fraction of treated cells, demonstrating progressive release of cathepsin in the cytosol. (f) The green fluorescent protein corona of PS-NH2 NPs is trafficked to the lysosomes and is degraded, while the lysosomes are swelling and become permeable, allowing cytosolic release of cathepsins. Adapted from Wang et al. [32] with permission
The different concentrations of intracellular proteins, nutrients, and ions also destabilize nanoparticles and set on their aggregation [14]. The aggregation might also be triggered after cellular enzymes have stripped away the protein corona and the particles’ organic coating [33]. The aggregation phenomenon is the principal cause that might affect the physical properties , implicated in imaging protocols [34]. Conversely, nanoparticles’ interactions with different cellular components might also result in a rearrangement—sorting and individualizing the nanoparticles in the periphery of endo-lysosomic compartments, in order to set on or accelerate particle degradation [35].
Finally, the cells are not only able to internalize the particles, but can also, under specific conditions, release the particles to adjacent or distal cells of the same or different types [36]. Particles’ expulsion via cell microvesicles should be taken into account in protocols involving highly sensitive modalities, such as high-field MR scanners or scanners provided with cryogenically cooled MRI probes, which are currently used mainly in preclinical cellular MRI [37].
1.6 Different Aspects of Biotransformation: Therapeutic Properties, Loss of Functionality, and Potential Toxicity
Bio-interactions and bioprocessing of the particle are the sine qua non of a particle’s life cycle within the organism. Particles’ surface (enriched with synthetic or biologically acquired components, such as the protein corona ) governs particles’ early biodistribution in the short run [35]. Nevertheless, once the biomolecules and parts of the synthetic coating are stripped off, the remnants of the synthetic coatings, particles’ rearrangement within cellular compartments, and the nature of the core start governing the long-term persistence within the organism, and the degradation and the elimination processes [29, 35, 38]. The biotransformations of nanoparticles, as well as their potential toxicity, tightly depend on the particles’ physical and chemical properties. Yet, some common features characterize the fate of inorganic nanoparticles regardless of their composition, structure, and size. Engineered nanoparticles are mainly captured by macrophages of the liver and spleen and thus tightly packed in their endo-lysosomes, which induce adsorption of biomolecules, particles’ aggregation, and gradual erosion of the coating and inorganic core, affecting in turn the physical and chemical properties that are required for imaging or therapeutic protocols [34, 39]. In addition, the degradation and processing might alter the toxicological profile of the administered material [40], and what used to be a small benign object now starts releasing its chemical (molecular), mainly metallic, components (ions), which are more or less rare in the organism and might trigger a cascade of adverse reactions, as the ones induced by the generation of reactive oxygen species (ROS) [41, 42]. These molecules might, under certain conditions, alter the physiological processes of the cell by inducing the lipid peroxidation of the cell membrane, set on mitochondrial and lysosomal damage, or damage the endoplasmic reticulum, which may result in programmed cell death. Nevertheless, antioxidant enzymes (such as superoxide dismutase, glutathione peroxidase, catalases), vitamins (such as α-tocopherol), or hormones (such as melatonin) are some of the antioxidant molecules present in biological systems that are capable of scavenging the ROS [43]. Be that as it may, “sola dosis facit venenum” (i.e., the dose makes the poison) [44] and the cooperation between the gradual occurrences of chemical factors (e.g., gradual dissolution of inorganic nanoparticles and progressive reduction of ROS by local antioxidants) and physical (e.g., aggregation and thus delayed degradation by hindering the access of biomolecules to particle surface) and biological phenomena (e.g., up-regulation of genes coding for specific proteins) [45] accompanying particles’ degradation over time might counterbalance or moderate the impact of the foreign materials’ burden to cells/organisms. Thus, in order to assess the factual toxicity of nanoparticles, their biotransformations have to be monitored under dynamical conditions over relevant periods of time. In addition, the conditions under which some inorganic nanoparticles might have an increased toxicity due to accelerated dissolution, such as UV illumination of QDs [40], should be considered and, if appropriate, avoided.
1.7 Material-Specific Degradation Patterns of Inorganic Nanoparticles In Vivo and Over Time
The specific chemical composition of the nanoparticle’s core unconditionally determines nanoparticle’s degradation pathways. Organisms possess a large variety of proteins, including enzymes, which are involved in the biotransformation of xenobiotics . As biotransformation allows the elimination or recycling of nanoparticle components, the progressive processing of a crystal to ions might alter the toxicological profile of the administered material.
While different degradation patterns are certainly intuitive, empirical evidence, obtained from gold/iron oxide heterodimers [38] or silver/gold nanohybrids [46], is particularly conclusive. The follow-up of the fate of gold/iron oxide heterodimers [38] evidenced the evolution of the ratio of magnetic iron over gold content and highlighted the distinct distribution and fate of gold and iron oxide within the organs. Over time the ratio was diminishing in the liver, while it remained almost constant in spleen, reflecting the prominent degradation of iron oxide in the liver. Spleen preferentially accumulates gold and is less efficient to degrade both materials. A fraction of heterodimers was able to fragment to individual gold or iron oxide subunits at early time points and the separated subunits had distinct distribution paths. Interestingly, such element-specific biodistribution has also been observed for NIR-absorbing hollow Ag-Au nanoshells, used for photothermal therapy in tumor-bearing mice [46]. The Ag/Au ratio varied considerably between organs, suggesting a size-dependent fragmentation of the heteroparticles at some time point post-injection. Gold was more present in liver and spleen, whereas Ag predominated in tumors for the smallest particles of 43 nm. The thinner shell allowed the 43 nm particles to fragment quicker and release Ag from the core-shell particles. This study again illustrated the different fate of materials and the importance of architecture on the life cycle of particles. Another important aspect is that the fate of NPs used for diagnostic or therapy should be investigated also after stimulation (i.e., laser exposition for photothermal ablation), as the external stimuli may also induce structural transformations of the particles.
2 Iron Oxide Nanoparticles
IONPs are mostly made of magnetite (Fe3O4) and/or maghemite (γ-Fe2O3) nanocrystals, which may exhibit different shapes (spheres, cubes, rods, rings, flowers, pyramids, octapods, nanoworms, etc.). These IONPs have superparamagnetic properties, and thus might be used in magnetic resonance imaging (MRI) or magnetic particle imaging (MPI) . The IONPs might be applied either systemically by direct application (e.g., intravenous injection) of the suspension of nanoparticles or by the administration of heterologous or homologous cells, which were previously labeled in vitro with IONPs [47]. In addition to the imaging functionality, these nanoparticles could potentially be used for cancer treatment, either for magnetic targeting [48] or magnetic hyperthermia [49], or as magnetothermal effectors for deep brain stimulation [50].
2.1 Role, Effect, and Fate of IONP Synthetic (Polymer) and Biological (Corona) Coating
IONPs are commonly coated with organic polymers, such as polyethylene glycol (PEG), oleic acid, dextran, chitosan, phospholipids, and other amphiphilic polymers. The latter increase the stability of IONP suspensions and increase the circulation time in blood after intravenous administration. Administered nanoparticles’ surfaces become covered by proteins [10], which are either bound to the synthetic coating or, if the polymer is stripped off, adhere directly to the particles’ core.
Magnetic IONPs and protein associates can be specifically probed with a method, which relies on the particles’ potential of orienting in the direction of the magnetic field [51]. When the magnetic field is switched off, the particles disorient and their Brownian relaxation time is proportional to their hydrodynamic volume. This relaxation time can be determined by the measurements of the magnetically induced optical birefringence signal. Combined to magnetic sorting and protein electrophoresis, the measurements of the magnetically induced optical birefringence signal [51] confirmed that protein adsorption strongly depends on surface coating and that the composition of the corona differs when IONPs are incubated in a medium supplemented with 10 % of plasma (generally used for cell culture) or when particles are suspended in pure plasma, which resembles in vivo conditions (Fig. 4). Moreover, some proteins, namely albumin and apolipoprotein, have a stabilizing (“individualizing”) effect on the nanoparticles, while other proteins, such as fibrinogen, trigger particles’ aggregation [51]. Remarkably, the particles with different protein corona not only exhibit different physical states (single particles or aggregates), but can also coexist in plasma and are differently processed by immune cells. Macrophages will neither react nor capture these populations in the same manner or time course. This experiment clearly exemplifies how interactions between biomolecules and nanoparticles can drastically influence the aggregation pattern, distribution, and fate of intravenously injected IONPs [51].

Fig. 4
Nanoparticles’ interactions with blood proteins in vivo and in vitro. The adsorption of proteins on magnetic nanoparticles can be determined specifically by measurements of the relaxation of the magnetically induced optical birefringence of magnetic nanoparticles’ suspensions [51], which reveals different populations of particles with distinct biological (nature of protein corona) and physical states (aggregation) that interact differently with macrophages
While polymer coatings increase the stability of nanoparticles, it has also been shown that they impact the long-term degradation of IONPs , as PEG-coated IONPs degraded more rapidly in comparison to amphiphilic polymer-coated IONPs, monitored over 1 year [38]. Nevertheless, as we emphasized earlier in this chapter, studies also show that once applied, the nanoparticle core and the polymeric shell dissociate [25, 28]. While the particles remain in the liver and spleen, some components of the coating can be eliminated in urine [28].
2.2 Cell Internalization of IONPs and Its Effects on Nanoparticles’ Magnetic Properties
Particle destabilization can be due to the loss of the synthetic coating, the adsorption of the host’s biomolecules, or arising from active biological processes, such as cellular internalization. Intracellular traffic together with the fusion and fission of endocytic compartments leads nanoparticles to lysosomes , where degradation occurs. As most nanoparticles, IONPs eventually reach the lysosomes of hepatic or splenic macrophages.
The subcellular confinement affects the physical properties of magnetic IONPs. At short distance between each other, IONPs experience dipole-dipole magnetic interactions and have less freedom to rotate and translate. This directly impacts the dynamical superparamagnetic behavior of IONPs: the magnetic susceptibility starts to decrease while the magnetic hysteresis (remnant magnetization) and blocking temperature (transition between superparamagnetic and ferromagnetic behavior) increase [52]. Intracellular confinement nevertheless has its advantage for cell detection by MRI [34], as the cell that confines a large amount of magnetic sources in a limited volume highly affects the homogeneity of the magnetic field. The local inhomogeneity thus results in MR signal loss that co-localizes with magnetically labeled cell. Once a cell population was magnetically labeled, the cells’ migration can be noninvasively monitored over time, particularly for the evaluation of the efficiency of cell therapy [53, 54].
Magnetic characterization of biological tissues allows obtaining the information on local confinement of magnetic nanoparticles within different organs [55]. Furthermore, magnetic measurements are consistent with morphological findings observed by TEM, which show that lysosomes of hepatic/splenic macrophages concentrate a higher number of nanoparticles than the lysosomes of circulating monocytes/macrophages that can be recruited to sites of inflammation [52]. The follow-up of nanoparticles’ magnetic properties also provides important information on nanoparticles’ biotransformation within organs, which directly impact those nanoparticles’ properties that are necessary for therapeutic and diagnostic applications.
2.3 Degradation and Recycling of IONPs
In order to characterize the degradation of administered nanoparticles over time in the body, we require specific methods of quantification, which allow detecting an infinitesimal fraction (ppm or ppb) of material per kg of total body weight (bw). Ideally, the method of quantification should permit to distinguish between nanoparticles and their degradation products.
The quantification or the evaluation of the distribution of IONPs and their degradation products in vivo is complicated by the quantity of endogenous iron (about 50 mg/kg b.w.), which is greater than the amount of iron that is administered for MRI (typically 2.5 mg/kg). Consequently, elemental analysis is inappropriate for IONP quantification. On the other hand, methods based on nanomagnetism such as electron paramagnetic resonance (EPR) allow us to specifically and sensitively quantify IONPs [45]. Measurements of magnetization as function of strength of magnetic field or temperature may supplement the data obtained by EPR , as they provide information about particles’ size and aggregation state. Besides, MRI provides a mean for monitoring IONPs’ distribution over time in the same animal, although quantification remains challenging, since the effect of IONPs on MR relaxivity depends on the local environment and physical state of particles. Multiscale and pluri-method scanning should therefore be used in order to track the distribution and transformation of IONPs.
Different methods have been proposed to monitor the degradation of IONPs with different shape, organization, and surface coatings. Some of them included 7–8 nm spherical IONPs with a hydrophilic glucose-derivative coating; 20 nm IO nanocubes, coated with polyethylene glycol (PEG) (Fig. 5); and 20 nm hetero-structures (dimers) made of gold/iron oxide, coated with an amphiphilic polymer or PEG. Following intravenous administration (2.5 mg/kg b.w.), IONPs mainly reach the liver and, to a lesser extent, the spleen. As expected, the nature of IONP coating determined the initial uptake in the reticuloendothelial organs [45, 49]. The quantity of superparamagnetic iron diminished with time, more rapidly in the liver than in the spleen, as evidenced by MRI and EPR analysis. A potential translocation/elimination of intact particles out of the organs, together with nanoparticle degradation within the organs, account for the superparamagnetic property loss, which results in a decrease of magnetic iron [45].

Fig. 5
Intracellular degradation of PEG-coated IONPs in murine liver and spleen after intravenous application, evidencing the transfer of iron into ferritin proteins. The intravesicular distribution of nanoparticles evolves over time: early after injection (day 1), dense assemblies of nanocubes avoid the cell from particle degradation and release of cytotoxic free iron ions. At longer times (days 7 and 14), the nanoparticles are transferred into protein-rich lysosomes in which they are more dispersed and susceptible to degradation. Red arrows show the coexistence of monodisperse iron-rich ferritin protein of 5.3± 0.8 nm diameter with some degraded or resilient nanocubes, suggesting a protein-triggered degradation and local iron transfer (a). High-resolution imaging performed 14 days after injection shows that degraded and resilient nanocubes maintain their initial crystalline structure (the spinel inverse and the vacancy-ordered γ-Fe2O3 structures along the [103] and [116] zone axes on the images (b, d), respectively). The blue arrows highlight the rough surfaces of the degraded nanostructures. The Fourier transform of the atomic structure of the ferritin core in image (b) (spleen day 14 post-injection, red squares) shows unambiguously a hematite structure oriented along the [481] zone axis. Adapted with permission from Lartigue et al. [35]
In complement to macroscopic findings, transmission electron microscopy (TEM), electron diffraction, and associated spectroscopy provide information about the evolution of the subcellular distribution and morphology of IONPs over time after intravenous injection. Interestingly, while nanoparticles are mainly found as clusters within lysosomes of splenic and hepatic macrophages 1 day post-injection, particle sorting was observed in the periphery of lysosomes or in electron-dense areas of the lysosomes at later time points (Fig. 5) [35]. Nanoscale observations also showed that IONPs become increasingly surrounded by monodisperse electron-dense nano-sized (6 nm) objects, which are rich in iron, but poorly crystalline (Fig. 5) [35, 45]. These nanostructures, also known as ferritin , an endogenous iron-storing protein or apoferritin, when not combined with iron, consist of 24 protein subunits that form a hollow shell, where a maximum of 4500 iron atoms can be stored in mineral crystallites of iron oxyhydroxide (mainly ferrihydrite) and other forms of iron oxide. Ferritin binds iron ions and regulates the occurrence of Fenton’s reactions, in which ferrous ions react with hydrogen peroxide and form reactive oxygen species that would damage the cells. Iron homeostasis is a finely regulated process where sets of proteins detect, transport, regulate, and store the metal, in order to provide it to the body at its demand (i.e., for hemoglobin synthesis) [56]. The coexistence of ferritins situated proximally to IONPs suggests a mechanism of local iron transfer from degraded IONPs to endogenous iron-storing proteins (Fig. 5). Although the morphological degradation of polydisperse 7-8 nm spheres is difficult to ascertain in vivo , nanocubes and gold/iron oxide heterostructures show evident features of erosion in liver and spleen 7 days after administration.
While the magnetic iron decreases, the nonmagnetic iron (total iron measured by elemental analysis subtracted by superparamagnetic iron measured by EPR ) increases, confirming the local transformation of superparamagnetic particles into nonmagnetic iron species. In addition, as the magnetic signature of IONPs fades away due to degradation, the low-temperature magnetic signature for ferrihydrite appears [45]. The local degradation of IONPs and iron transfer in ferritins occurs within the lysosomes on nanoparticles’ aggregate margins, where the iron remains bound in crystals until the proteins are synthesized and recruited in the vicinity of nanoparticles. Studies performed more than two decades ago have evidenced that IONP degradation is followed by the recycling of IONP compounds [23, 24]. The labeling of IONP core with 59Fe showed that the iron from NPs incorporated within the hemoglobin of red blood cells in rats [23] and mice [24].
2.4 In Vivo Toxicity of IONPs
IONPs are generally considered as biocompatible, safe, and nontoxic. The median lethal dose (LD50) of intravenously applied citrate-coated ultrasmall (8.6 nm) IONPs in mice was reported to be greater than 949 mg (17 mmol) Fe/kg body weight (b.w.) [57], which is almost 400 times more than what is required for MRI imaging . For comparative purposes note that the LD50 of cisplatin, a commonly used anticancer therapeutic agent, is 11 mg/kg b.w. after i.v. injection in mice [58]. Indeed the LD50 values of IONPs vary in other species, when other coating agents are used or when IONP cores and/or hydrodynamic sizes differ. As an example, the LD50 of (8 ± 3 nm) IONPs after i.v. administration to rats was 36.42 mg/kg b.w. [59].
As the liver (either due to the phagocytic activity of its macrophages , the Kupffer cells, or due to specific ligands that target hepatocytes) remains the main accumulation organ after administration of most types of IONPs , the administration of such nanoparticles, which may induce oxidative stress, might result in an increased level of liver enzymes. Several studies published thus far reported on low or minor transient changes in liver enzyme levels after dextran- and pluronic-coated IONPs (dose 10 mg iron/kg b.w.) [60]. The administration of dextran-coated IONPs to humans did not induce any clinically significant adverse reactions.
3 Quantum Dots
QDots are semiconductor nano-crystallites typically made of lead sulfide, lead selenide, cadmium selenide, cadmium telluride, cadmium sulfide, indium arsenide, or indium phosphide that are emerging as potential fluorescent probes for biomolecular and cellular imaging. Due to their outstanding brightness, high photostability, and broad absorption spectrum coupled with size-dependent narrow emission bands, QDots have been particularly attractive as efficient multiplexing probes for cell labeling and tracking. Their potential biomedical applications mainly concern in vivo imaging for early cancer detection. Unlike conventionally used organic dyes and fluorescent proteins, QDots have very large molar extinction coefficients, on the order of 0.5–5 × 106/M/cm which is 10–50 times greater than those of organic dyes [2], and could thus allow sensitive detection of analytes at a lower concentration, which is especially important for detection of cancerous lesions at their early stage. In order to efficiently fluoresce, QDot cores should be protected with a shell. The latter can be made of ZnS, silica, or different polymers.
3.1 Cellular Processing and Fate of QDots
The optical properties of QDots are affected by cell internalization : agglomeration into endosomes and lysosomes , as well as intralysosomal acidic pH. These changes in overall fluorescence of the QDots complicate the quantification of intracellular QDots . For example drastic reductions in QDot photoluminescence quantum yield may result from oxidation or surface defects upon exposure of QDots to low pH. Oxidative dissolution of the QDots and rapid reduction in fluorescence emission have been observed despite the presence of organic shells on the surface of the QDots [40].
From the biological point of view, persistent localization of CdTe-based QDots in lysosomes might result in an enlargement of the lysosomal compartment as an adaptive response to the presence of QDots, and an enhancement of the antioxidant response mediated by intracellular glutathione. Activation of the transcription factor TFEB, a key regulator of lysosome biogenesis , shows that cells are able to sense the overload of lysosomes with nanoparticles and adapt their response to diminish oxidative stress [61].
Pioneering studies [62, 63] used either the fluorescence emission of QDots or the elemental quantification of cadmium to track QDots ex vivo and report on their particokinetics and biodistribution. Long-term experiments demonstrated that the QDots remained fluorescent after at least 4 months in vivo suggesting that they keep their integrity [63]. Fischer et al. observed the sequestration of lysine-coated and BSA-coated CdSe/ZnS in RES cells, but no short-term excretion in urine and feces, nor degradation of the inorganic part of QDots [62]. To assess degradation, they used ultracentrifugation on the digested organs to separate the intact QDots in tissue from isolated ions. Fitzpatrick et al. investigated the 2-year fate CdSe/ZnS Qdots coated with PEG 5000 intravenously injected in mice [64]. The fluorescence emission of QDots was observed in the blood immediately after injection and later in the liver, spleen, lymph, and bone marrow. However, the emission in liver faded after 5 days. Signal fade-out was slower in the bone marrow (3–6 months) and even slower in lymph nodes (up to 2 years). It was not clear whether this fading was due to particle dissolution, darkening, or excretion mechanisms. Surprisingly, two-photon spectral confocal microscopy indicated the presence of QDot clusters in liver, spleen, and lymph nodes 2 years post-injection, but their fluorescence was significantly blue-shifted [64], which differs from previous reports reporting red-shifting attributed to QDot clustering. Interestingly, the blue shift in peak position was not seen at early time points, neither attributed to storage and was due solely to the long residence time in living animals, indicating partial degradation of the semiconductor crystals (Fig. 6). In spite of the ZnS shell and polymer coating, the blue shift could be due to the loss of core materials, a change in the size/shape ratio of the nanocrystal, or the degradation of the ZnS shell. The absence of apparent toxicity of QDots , injected at a concentration that would show lethal toxicity as soluble cadmium ions (8.5 mg/kg of Cd, equivalent to about two lethal doses of Cd2+ injected intravenously), and the persistent fluorescence signal indicate that some particles remained almost intact after 2 years without substantial leakage of cadmium, but that they had experienced some crystal or surface modification explaining the blue shift. In the mentioned study, nanoscale examination of the morphology and elemental composition of the persistent particles would be necessary to provide insight into the long-term biotransformation of QDots.