1. Pulmonary arterial hypertension (PAH)
1.1. Idiopathic
1.2. Heritable
1.2.1. BMPR2
1.2.2. ALK1, endoglin (with or without hereditary haemorrhagic telangiectasia)
1.2.3. Unknown
1.3. Drugs and toxins induced
1.4. Associated with (APAH)
1.4.1. Connective tissue diseases
1.4.2. HIV infection
1.4.3. Portal hypertension
1.4.4. Congenital heart disease
1.4.5. Schistosomiasis
1.4.6. Chronic haemolytic anaemia
1.5. Persistent pulmonary hypertension of the newborn
1′. Pulmonary veno-occlusive disease and/or pulmonary capillary haemangiomatosis
2. Pulmonary hypertension due to left heart disease
2.1. Systolic dysfunction
2.2. Diastolic dysfunction
2.3. Valvular disease
3. Pulmonary hypertension due to lung diseases and/or hypoxia
3.1. Chronic obstructive pulmonary disease
3.2. Interstitial lung disease
3.3. Other pulmonary diseases with mixed restrictive and obstructive pattern
3.4. Sleep-disordered breathing
3.5. Alveolar hypoventilation disorders
3.6. Chronic exposure to high altitude
3.7. Developmental abnormalities
4. Chronic thromboembolic pulmonary hypertension
5. PH with unclear and/or multifactorial mechanisms
5.1. Haematological disorders: myeloproliferative disorders, splenectomy
5.2. Systemic disorders: sarcoidosis, pulmonary Langerhans cell histiocytosis, lymphangioleiomyomatosis, neurofibromatosis, vasculitis
5.3. Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders
5.4. Others: tumoural obstruction, fibrosing mediastinitis, chronic renal failure on dialysis
Classification of PH in CHD
The majority of PH in congenital heart disease (CHD) comes under the umbrella of pulmonary arterial hypertension (PAH, Group 1 in the WHO Classification) when it is referred to as PAH associated with CHD (APAH-CHD) (Table 25.1). Being ‘pre-capillary’ PH, the pulmonary wedge pressure is ≤15 mmHg with pulmonary vascular resistance >3 Wood units, and the cardiac output is normal or reduced in the absence of an alternative explanation, i.e. lung disease (Group 3), chronic thromboembolic PH (Group 4), or PH associated with unclear/multifactorial mechanisms (Group 5) [1].
Shunts are the commonest CHD abnormality associated with PAH, making up approximately 38 % of patients with CHD and PH in one cohort [2]. This group includes Eisenmenger’s syndrome, where the development of PH leads to reversal of blood flow through the shunt and cyanosis, but primarily involves patients with systemic-to-pulmonary shunts and to a lesser extent those with small defects and PAH post-corrective surgery (Table 25.2). A helpful five-point framework by which these individual lesions can be characterised has been provided by the most recent guidelines [1] (Table 25.3).
Table 25.2
Classification of shunts in APAH-CHD
(a) Eisenmenger’s syndrome |
Eisenmenger’s syndrome includes all systemic-to-pulmonary shunts due to large defects leading to a severe increase in PVR and resulting in a reversed (pulmonary to systemic) or bidirectional shunt. Cyanosis, erythrocytosis and multiple organ involvement are present |
(b) Pulmonary arterial hypertension associated with systemic–to–pulmonary shunts |
In these patients with moderate to large defects, the increase in PVR is mild to moderate, systemic-to-pulmonary shunt is still largely present, and no cyanosis is present at rest |
(c) Pulmonary arterial hypertension with small a defects |
In cases with small defects (usually ventricular septal defects <1 cm and atrial septal defects <2 cm of effective diameter assessed by echocardiography), the clinical picture is very similar to idiopathic PAH |
(d) Pulmonary arterial hypertension after corrective cardiac surgery |
In these cases, congenital heart disease has been corrected, but PAH is either still present immediately after surgery or has recurred several months or years after surgery in the absence of significant post-operative residual congenital lesions or defects that originate as a sequela to previous surgery |
Table 25.3
5-point framework with which to classify shunts in APAH-CHD
1. Type |
1.1. Simple pre-tricuspid shunts |
1.1.1. Atrial septal defect (ASD) |
1.1.1.1. Ostium secundum |
1.1.1.2. Sinus venosus |
1.1.1.3. Ostium primum |
1.1.2. Total or partial unobstructed anomalous pulmonary venous return |
1.2. Simple post-tricuspid shunts |
1.2.1. Ventricular septal defect (VSD) |
1.2.2. Patent ductus arteriosus |
1.3. Combined shunts |
Describe combination and define predominant defect |
1.4. Complex congenital heart disease |
1.4.1. Complete atrioventricular septal defect |
1.4.2. Truncus arteriosus |
1.4.3. Single ventricle physiology with unobstructed pulmonary blood flow |
1.4.4. Transposition of the great arteries with VSD (without pulmonary stenosis) and/or patent ductus arteriosus |
1.4.5. Other |
2. Dimension (specify for each defect if more than one congenital heart defect exists) |
2.1. Haemodynamic (specify Qp:Qs)a |
2.1.1. Restrictive (pressure gradient across the defect) |
2.1.2. Non-restrictive |
2.2. Anatomicb |
2.2.1. Small to moderate (ASD ≤2.0 cm and VSD ≤1.0 cm) |
2.2.2. Large (ASD >2.0 cm and VSD >1.0 cm) |
3. Direction of shunt |
3.1. Predominantly systemic to pulmonary |
3.2. Predominantly pulmonary to systemic |
3.3. Bidirectional |
4. Associated cardiac and extracardiac abnormalities |
5. Repair status |
5.1. Unoperated |
5.2. Palliated [specify type of operation(s), age at surgery] |
5.3. Repaired [specify type of operation(s), age at surgery] |
The likelihood of developing PAH depends on the shunt itself – in patients with pre-tricuspid defects, it can be rare (i.e. ASD) or up to 15–20 % for sinus venosus defect. For post-tricuspid defects, it is more common (50–70 % for large VSD/PDA) and almost universal in patients with truncus arteriosus [3]. It is important to recognise that not all PH in CHD is due to PAH. Other causes include left heart disease (Group 2) and lung disease (Group 4), including obstructive sleep apnoea.
Pathophysiology and Presentation
In CHD, the pulmonary vasculature is exposed to higher than normal blood flow (i.e. pre-tricuspid shunts) and pressures (in post-tricuspid shunts). This leads to damage of the vascular endothelium which sets into motion a series of deleterious pathways. These culminate in a pulmonary vasculopathy characterised by medial hypertrophy and plexiform lesions. The end effect is elevated pulmonary vascular resistance corresponding to an attenuation of pulmonary vasculature.
Symptoms of PH tend to be non-specific and include breathlessness, effort intolerance, chest pain and syncope. Presentation is influenced by a number of factors including the severity of PH, the nature of the underlying defect and prior surgery. PH associated with post-tricuspid shunts tends to manifest with symptoms in infancy whilst pre-tricuspid shunts may take a number of decades to become apparent.
Clinical features are dominated by those of PH (i.e. elevated venous pressure, right ventricular heave, loud second heart sound, ascites, peripheral oedema) with diminishment of murmurs associated with post-tricuspid shunts with the onset of the Eisenmenger’s syndrome [4].
Epidemiology and Outcomes
Consistent data has emerged from contemporary registries defining the frequency with which PH affects patients with CHD. In the Dutch National Registry, PH (defined as an echocardiographic-derived systolic PA pressure >40 mmHg) affected 5 % of adults with CHD [5] compared to data from Quebec, Canada, where 5.8 % had received a diagnosis of PH [2].
When considered in terms of etiologies underling PAH, CHD is an important contributor (Fig. 25.1). In the REVEAL Registry of consecutively screened PAH patients, the most common individual subset was idiopathic PAH (46.2 %), followed by collagen vascular disease/connective tissue diseases (25.2 %) and then CHD (9.8 %) [6].
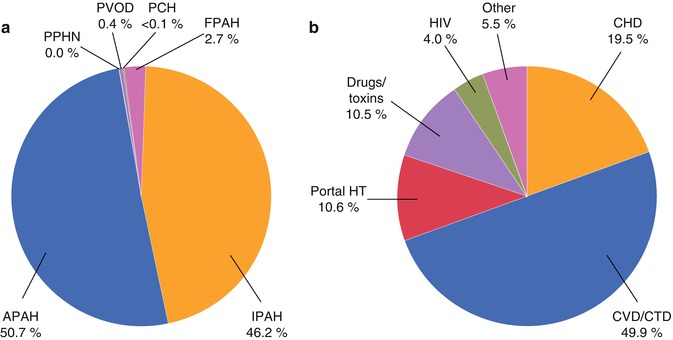
Fig. 25.1
Pie charts demonstrating percentage of patients with WHO Class I (PAH, a) and of these the percentage comprising associated PAH in the REVEAL Registry (b). Note that CHD is the third most common diagnosis underlying PAH. APAH associated PAH, CHD congenital heart disease, CVD/CTD colleague vascular disease/connective tissue disease, FPAH familial PAH, HT hypertension, IPAH idiopathic PAH, PAH pulmonary arterial hypertension, PCH pulmonary capillary haemangiomatosis, PPHN pulmonary hypertension of the newborn, PVOD pulmonary veno-occlusive disease, REVEAL Registry to Evaluate Early and Long-Term PAH Disease Management (From Badesch et al. [6], with permission)
It is not felt likely that these figures will remain static in the future [4]. Advances in surgical and medical treatments continue to expand the numbers of patients with CHD surviving to adulthood. This population is likely to comprise increasingly complex disease with higher rates of associated PAH.
As well as defining its prevalence, registry data has proven what was long suspected that PH in CHD confers a worse prognosis. In the Dutch group, patients with PH had a HR of 3.8 (95 % CI 2.7–5.4, p < 0.001) for all-cause mortality, adjusted for age and gender. In the Canadian cohort, patients with PH and CHD had a 2-fold higher all-cause mortality and 3-fold higher morbidity [2] than case controls without PH (Fig. 25.2). Finally, the REVEAL Registry has shown that children with APAH-CHD have a similar 5-year survival from RHC (71 ± 13 %) than patients with IPAH (75 ± 7 %) [7]. This argues against the assertion that patients with APAH-CHD fare better than those with IPAH since the RV in CHD has been ‘trained’ to deal with high right-sided pressures over a long period of time.
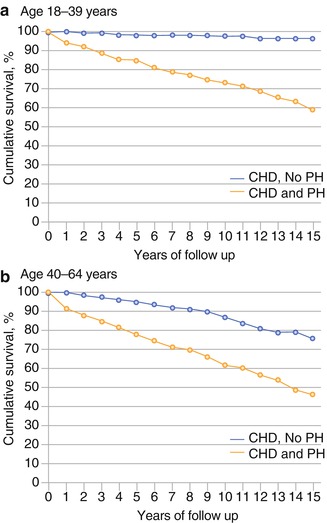
Fig. 25.2
Survival curves for adults with CHD without (triangular data points) and with PH (diamond data points) showing adverse prognosis in both 18–39-year-olds (a) and 40–46-year-olds (b) (From Lowe et al. [2], with permission)
Treatment
Interventions to the defect itself, when pulmonary blood flow is high but pulmonary vascular disease minimal, represent an important treatment goal in patients with APAH-CHD. The general consensus is that this should be undertaken when the pulmonary vascular resistance is less than 6 Wood units. Management is less clear when the value lies between 6 and 10 Wood units [3]. In Eisenmenger’s syndrome, shunt closure is contraindicated.
For the remainder of patients with APAH-CHD in whom an intervention is not possible, benefit has come in the wake of trials of targeted medical therapies in individuals suffering with idiopathic PAH. Agents include intravenous epoprostenol, endothelin receptor antagonist and phosphodiesterase-5 inhibitors which address deficiencies in prostacyclin (potent vasodilator) and overexpression of endothelin-1 and phosphodiesterase-1 (vasoconstrictors), respectively. Class I support for Bosentan and Class IIa for sildenafil and prostanoids has been given in the most recent recommendations [1].
The Role of Imaging in Management of PH in Congenital Heart Disease
Diagnosis
Given the potential to cure or prevent progression of PH, there is an imperative to identify it promptly in patients with established CHD and, conversely, to diagnose CHD when present in patients with a known diagnosis of PH. The recommended diagnostic algorithm [1] for patients suspected of having PH focuses first on exclusion of common causes of PH in order of likelihood (Groups 2 and 3 and then chronic thromboembolic PH with a VQ scan). If these investigations are non-contributory, a RHC is then recommended before specific tests are undertaken to identify rarer causes of PH including CHD. For patients with established CHD, there is merit in taking the same systematic approach given the potential for there being more than one cause contributing to PH.
The algorithm emphasises the key role of multimodality imaging in patients with APAH-CHD, recognising the strengths and complementary nature of these modalities and the complex nature of the diagnostic questions arising.
Being portable, cheap and available transthoracic echocardiography is used first. It allows assessment of intracardiac shunts (site, size and direction) as well as identifying features which might suggest PH. These include right ventricular remodelling (dilatation, hypertrophy and impaired systolic function), right atrial enlargement, compression of the left ventricle, pulmonary artery dilatation and pericardial effusion. Flattening of the interventricular septum can also be quantified using the eccentricity index [8].
The extent of right ventricular dilatation is best appreciated in the apical four-chamber view. Right ventricular size can be quantified using linear dimensions, fractional area change, M-mode and tissue Doppler. Enlargement of the right atrium can be quantified. Right ventricular hypertrophy is measured in the subcostal view.
Right heart and pulmonary pressures can be non-invasively estimated using Doppler interrogation of the tricuspid regurgitant (TR) jet, right ventricular outflow tract acceleration time, pulmonary regurgitation jet and M-mode measurements of the inferior vena caval size and response to respiration. In day-to-day practice, TR jet velocity (V) is most often used. The right ventricular systolic pressure can be calculated by adding right atrial pressure to 4V [2]. This equates to systolic pulmonary artery pressure in the absence of pulmonary stenosis. PH is likely when velocity is >2.8 m/s particularly when a dilatation or dysfunction of the right ventricle coexist.
The ESC guidelines [1] draw attention to the finding of higher than expected pulmonary blood flow in the setting of a small shunt or significant PA dilatation with only moderate PH as these are strongly suggestive of an aberrant pulmonary vein or sinus venosus defect. In these cases, transoesophageal echocardiography can be particularly helpful in imaging the atrial septum, specifically where the superior vena cava enters the right atrium.
Cardiac Computed Tomography
Although disadvantaged by the need for ionising radiation and contrast, computed tomography (CT) has an important part to play in the investigation of APAH-CHD where it can provide information on cardiac chambers, great arteries, lung vasculature and parenchyma and mediastinal structures in a single acquisition with high spatial resolution.
This is particularly the case when acoustic windows have been poor (limiting echocardiography), lung disease is present and devices including pacemakers prevent CMR scanning.
The same features of cardiac remodelling associated with PH described previously can also be identified using cardiac CT. Communications between chambers can be visualised and the direction of shunt inferred by the direction of contrast. Although excellent at providing anatomical detail, it is generally not the modality of choice for providing functional data. If necessary, biventricular function can be assessed though this necessitates greater radiation exposure to capture information throughout the cardiac cycle.
The CT pulmonary angiogram is the test of choice to assess the proximal and distal pulmonary arteries non-invasively. In doing so, pulmonary artery dilatation can be identified as well as thrombus, which may form in situ due to sluggish blood flow rather than being embolised from the lower peripheries.
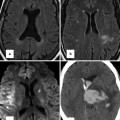
High-resolution CT scan provides valuable information on lung parenchyma which can be abnormal in patients with CHD because of bronchiectasis or hypoplasia. High-resolution CT may also detect parenchymal changes due to pulmonary arterial hypertension, for example, ground glass changes, nodular opacities and serpiginous intrapulmonary small vessels representing plexogenic arteriopathy (Fig. 25.3). In those with chronic thromboembolic PH, it may also identify within the lung tissue haemorrhage, infarction or a mosaic attenuation pattern (due to heterogenous lung perfusion). High-resolution CT has a vital role in identifying those with pulmonary veno-occlusive disease where advanced therapies might be harmful (Fig. 25.4). The key features of this rare entity on CT are interlobular septal thickening, ground glass shadowing and adenopathy.
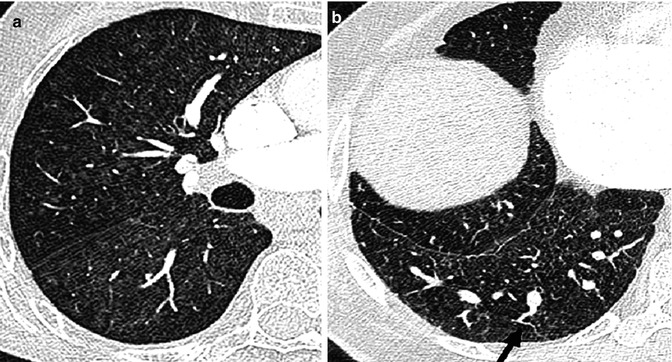
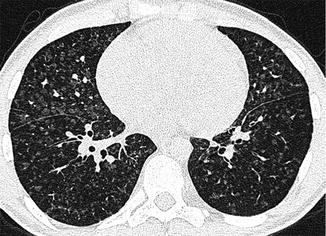
Fig. 25.3
High-resolution CT findings in a patient with pulmonary arterial hypertension. Ground glass changes and small nodular opacities are seen in the lung parenchyma in pulmonary arterial hypertension (a) and serpiginous intrapulmonary vessels (b), black arrow (Courtesy of Dr Michael Rubens, Royal Brompton Hospital, UK)
Fig. 25.4
High-resolution CT scan showing classic features of pulmonary veno-occlusive disease. Though this condition is rare, its recognition is important as conventional vasodilator treatment (Courtesy of Dr Michael Rubens, Royal Brompton Hospital, UK)
When contrast enhanced, this technique is particularly adept in the assessment of extracardiac features, particularly native or surgically fashioned systemic-to-pulmonary shunts. Collateral vessels are readily identified. These include dilated bronchial arteries (a commonly seen feature in PH) or bypassing vessels in cases of pulmonary venous occlusion.
Enlarged central pulmonary arteries is a non-specific sign and can be seen in patients with large left-to-right shunt with or without increased pulmonary vascular resistance. A cross-sectional diameter of >3 cm or larger than the aorta for distal main pulmonary artery is suggestive of pulmonary hypertension. Abrupt decrease in the calibre of segmental and subsegmental arteries may indicate increased pulmonary vascular resistance. An analytic technique called fractal analysis was employed to determine whether the degree of branching within the pulmonary arteries of children and young adults with PAH, half of whom had CHD (Fig. 25.5), could be used as a non-invasive measure of PH [9]. This index compared well with conventional markers of disease severity such as functional class, 6-min walk test distance and indexed pulmonary vascular resistance and predicted death amongst the cohort (HR 5.6 (95 % CI 1.2–25; p = 0.027)). Associated cardiac findings in CT may include enlarged right ventricle and atrium, bowing of the interatrial septum, straightening of the ventricular septum, right ventricle hypertrophy, dilatation and decreased fractional shortening of the right ventricle outflow tract and reflux of contrast into a dilated inferior vena cava, hepatic veins or azygos system.
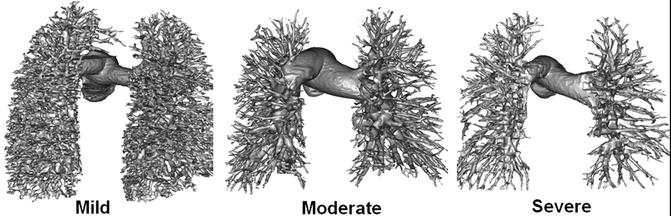
Fig. 25.5
Surface-rendered data taken from patients with increasingly severe PH (left to right). Note attenuation in branch pulmonary arteries. This is reflected in their fractal dimensions which diminish with reduced branching (1.66, 1.48 and 1.27, respectively) (From Moledina et al. [9] with permission)
Cardiovascular Magnetic Resonance
CMR has the strong advantage of being able to image in any plane with high spatial and temporal resolution without ionising radiation. As repeated examinations become commonplace, the latter characteristic is increasingly important.
In order to optimise image quality, breath holding is required which may be problematic in this population of patients. In addition, pacemakers/devices currently represent a contraindication to CMR.
CMR provides the gold standard assessments of right [10] and left ventricular size and function. Especially for the heavily trabeculated right ventricle, analysis remains dependent on the individual undertaking it.
Using both cine imaging and in-plane flow mapping, intracardiac shunts can be easily identified. By comparing the ratio of flow through the pulmonary artery to that in the aorta using flow mapping, Qp:QS can be determined and the shunt quantified.
With the addition of contrast, magnetic resonance angiography can delineate extracardiac shunts (especially partial anomalous venous drainage [11]) as well as the pulmonary vasculature.
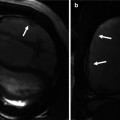
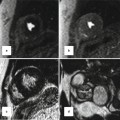
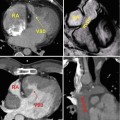
Late gadolinium enhancement typically demonstrates areas of enhancement at the RV-LV insertion points [12, 13]. Histological data however supports the concept that the myocardial disarray at these sites is a normal feature of insertion region anatomy exaggerated in PH by the hypertrophy of the right ventricle [14] (Figs. 25.6 and 25.7).
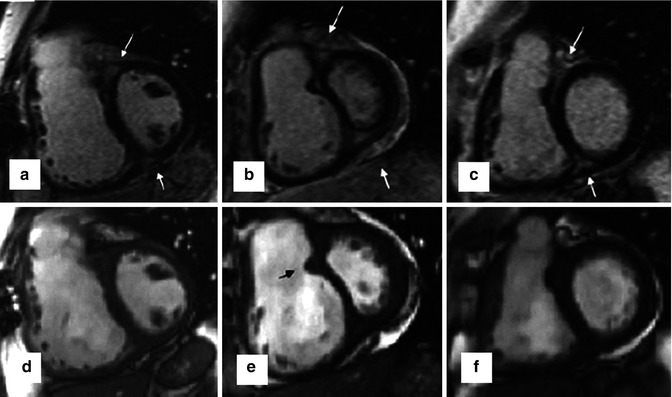
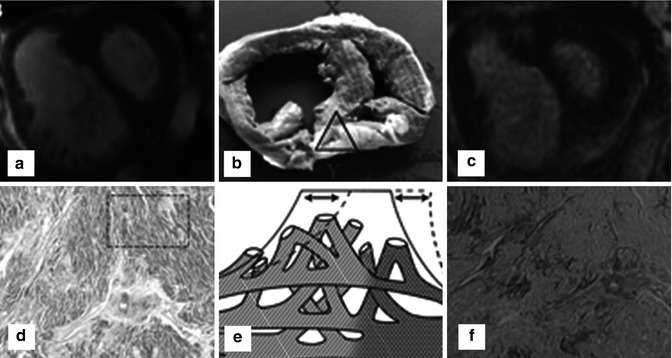
Fig. 25.6
The characteristic late gadolinium enhancement pattern of pulmonary arterial hypertension. The characteristic late gadolinium enhancement pattern of PH. (a–c) insertion region enhancement (arrows) is triangular in shape with the base at the epicardial surface where both ventricles meet and its apex directed into the interventricular septum. Corresponding short-axis cine slices (d–f). The septomarginal trabeculation is arrowed (e) – enhancement is often seen within this structure (From Bradlow et al. [15] with permission)
Fig. 25.7




Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
