Fig. 1
Schematic diagram of prostate carcinoma cell depicting transmembrane prostate specific membrane antigen (PSMA). The extracellular portion binds antibody J591 which has been used labeled with Lutetium-177 and other radionuclides and studied as a radioimmunotherapeuric agent. The intracellular portion of PSMA binds the antibody 7E11 which is the targeting vehicle for the diagnostic imaging agent, Prostascint®
2.1 PSMA as an Antigen Target for a Radiolabeled Antibody
PSMA itself has been isolated and a variety of antibodies have been developed to it. In 1998, capromab (7E11/CYT-356), a murine monoclonal IgG1 that binds to the intracellular portion of PSMA was labeled with Indium-111 [111In] and developed as a prostate carcinoma imaging agent. It was approved by the FDA in the United States and marketed as Prostascint ® [initially by Cytogen, Inc.] to evaluate extent of disease in high-risk patients presenting with Gleason scores of 7 or more and in patients with rising PSA following prostatectomy (Bartley et al. 2006; Kahn et al. 1998a, b; Jani et al. 2004a, b). Prostascint ® has not found widespread acceptance as an imaging agent in the urologic oncology community and efforts to utilize this antibody as a radioimmunotherapy agent have been unsatisfactory (Kahn et al. 1999; Deb et al. 1996). Since 7E11 preferentially recognizes the intracellular portion of the PSMA molecule, it is likely that localization depends upon the presence of necrotic cells. This conclusion is supported by observations in cell suspensions in which there is no demonstrable binding in viable LnCaP cells, but binding is demonstrated when cells are exposed to toxic or membranolytic reagents (Smith-Jones et al. 2000). Since the goal of a radiolabeled monoclonal antibody used for therapy is to destroy viable tumor cells, an epitope that is only available on necrotic cells represents a suboptimal target.
Subsequently, other monoclonal antibodies were developed (Liu et al. 1997). For more than a decade, our group has evaluated the therapeutic potential of an antibody identified as J591 that has a high degree of affinity for the external epitope of PSMA. Binding to viable prostate ca cells expressing PSMA has been demonstrated in cell suspensions [LnCaP cells], animal models of human prostate carcinoma, and other human tumors (Liu et al. 1997; Smith-Jones et al. 2003). After binding to the external epitope of PSMA, the membrane evaginates with internalization on the antigen–antibody complex.
The anti-PSMA monoclonal antibody, like other immunoglobulins, is readily iodinated but internalization of the antibody epitope resulted in enzymatic digestion of the immunoglobulin and release of small peptide fragments that diffuse from the cell reducing the tumor radionuclide residence time, and consequently the tumor radiation absorbed dose. Accordingly, it was decided to proceed with evaluation of the radiometals 90Yttrium [90Y] and 177Lutetium [177Lu] as potential labels for targeted radioimmunotherapy.
2.2 PSMA as an Enzyme Target for a Small Molecule Antagonist
Although incompletely understood, the complex PSMA molecule has an enzymatic function and a role in transmembrane transport. In recent years, it has been demonstrated that the small molecule glutamate-urea-lysine complex has a high degree of affinity for the external component of the complex PSMA molecule. In recent years, compounds based on the triplet molecule glutamate-urea-lysine have demonstrated binding to PSMA with high affinity and specificity (Chen et al. 2008) (Fig. 2). These observations have stimulated interest in the use of these compounds to target PSMA. After preliminary studies involving animal models bearing human prostate carcinoma demonstrating successful imaging of the implanted tumors, initial clinical imaging with an 123I–labeled glutamate-urea-lysine derivative metastatic prostate carcinoma in patients was performed (Hillier et al. 2009) (Fig. 3). The first of these compounds was iodinated with 123I for using as an imaging agent with an impressive degree of success (Fig. 4). The high affinity and specificity of the 123I-labeled glutamate-urea-lysine complex raised the possibility that a 131I-labeled molecule could potentially be used as a targeted radionuclide therapy agent. Subsequent studies in PSMA-expressing prostate cancer cells demonstrated that 131I-labeled versions of these urea-amino acid complexes were stable for long intervals (Babich J, personal communication; Haberkorn U, personal communication).
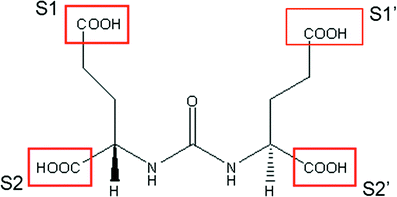
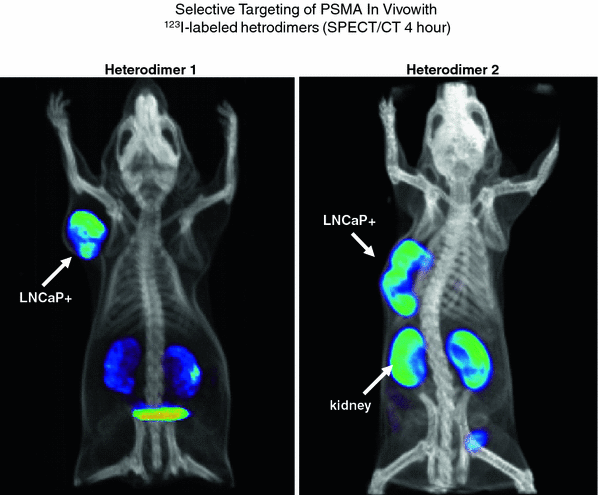
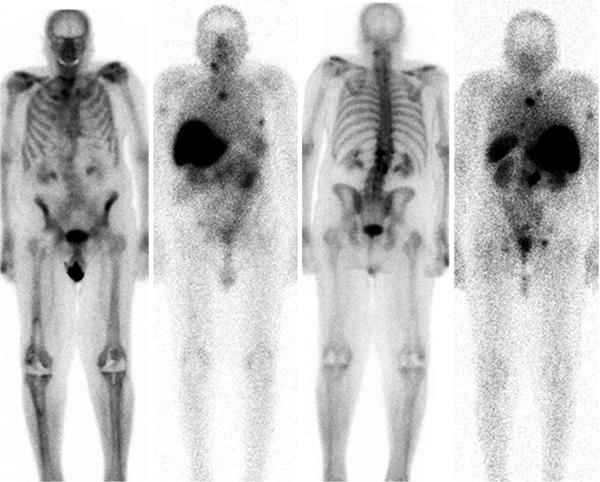
Fig. 2
Schematic diagram of the heterodimer glutamate-urea-glutamate which acts as an antagonist to the enzymatic function of PSMA (see text). S1 and S1’ can be modified to increase molecular weight and alter biodistribution. S1’ is tolerant of large hydrophobic groups without loss of affinity. S2 and S2’ are intolerant of structural changes. (Courtesy J. Babich, PhD)
Fig. 3
SPECT/CT of nude mice bearing PSMA+ prostate carcinoma in-plants. Two different heterodimers were labeled with I-123. Differences in the biodistribution and route of excretion are demonstrated. PSMA negative tumors as controls were also in-planted but are not visible
Fig. 4
Whole body scans, L to R-anterior bone scintigraphy and corresponding Lu-177 J591, 5 days post injection; posterior bone and Lu-177 J591 scans. Intense hepatic uptake is non-specific. Note activity in spleen, kidneys and modest GI tract contents. Several osseous metastases are identified based on imaging the gamma emission of Lu-177
When the external epitope of the PSMA moiety binds an antibody, the epitope-antibody complex is internalized and the iodinated amino acid within the immunoglobulin is labile and elutes from the cell (Smith-Jones et al. 2000). By contrast, either the high affinity enzyme antagonist molecules are not internalized but remain bound to the PSMA-expressing cells or 131I labeling of these amino acid-urea complexes produces an iodinated molecule that is more stable than the usual iodo-tyrosine-labeled immunoglobulin.
3 Preclinical Studies
3.1 Preclinical Studies with Radiolabeled Anti-PSMA Monoclonal Antibodies
In order to select a charged particle emitting radionuclide to be used in clinical radioimmunotherapy studies, several candidate radionuclides were evaluated for ease of labeling, affect on immunoreactivity, retention within the cell, and radiobiologic evidence of cytocidal effect. The physical properties and labeling chemistry of 131Iodine [131I], 90Yttrium [90Y], and 177Lutetium [177Lu] are summarized in Table 1. 131Iodine, of course, has a long history of effective use in the treatment of thyroid carcinoma and as a radio label in other radioimmunotherapy protocols. Radioiodine labeling techniques are well-known and it is possible to synthesize stable iodinated compounds that withstand enzymatic cleavage of the iodine if the iodine is inserted into double bonds of other than a tyrosine ring. High-specific activity 131I is readily available and relatively inexpensive. During the 1990s, 90Y had come to be considered the preferred radionuclide for therapy because of the β energy greater than that of 131I with consequently longer range in tissue. The short half-life of 90Y is also a potential advantage because of the greater radiobiologic effect of any given radiation dose delivered over a shorter interval (known as the dose rate effect). In the late 1990s, 177Lu became available (courtesy of the Missouri University Research Reactor (MURR)]. 177Lu, like 90Y, is a radiometal; hence if the radioimmunoglobulin is internalized, it is likely to remain intracellular. Although the chemistry of 177Lu and 90Y is somewhat similar, the physical properties (physical half-life, β energy and accompanying γ emission) of 177Lu are more similar to 131I than to 90Y. Several methods to prepare chemically stable radiometal-labeled immunoglobulins have been developed. It was decided to evaluate the stability and utility of DOTA (1,4,7,10-tetra aza cyclo dodecane-1,4,7,10-tetra acetic acid) as a complex to covalently bind to the immunoglobulin and serve as a chelating moiety for the metallic radionuclides. The DOTA moiety has a high affinity for both 177Lu and 90Y.
Table 1
Beta emitting radionuclides for therapy
131I | 90Y | 177Lu | |
|---|---|---|---|
Physical half-life (days) | 8.05 | 2.67 | 6.7 |
β− particles (MeV)Max | 0.61 | 2.280 | 0.497 |
Average | 0.20 | 0.935 | 0.149 |
Range in tissue (mm)Max | 2.4 | 12.0 | 2.20 |
Average | 0.4 | 2.7 | 0.25 |
Gamma emission (MeV) | 0.364 (81 %) | None | 0.113 (7 %) |
0.208 (11 %) | |||
Equilibrium dose constant (rad.g/hr) | |||
β− radiation | 0.389 | 1.9886 | 0.314 |
Gamma | 0.815 | None | 0.075 |
During preliminary studies using 111In DOTA-J591 as a surrogate for the radiometals 90Y and 177Lu, the affinity of murine J591 to LnCaP cells and the stability of the bound radiolabeled immunoglobulins were demonstrated in intact animals. Although J591 binds to an external epitope of PSMA, the epitope-antibody complex is internalized and likely digested in the intracellular environment. Nevertheless, 111In DOTA-J591 demonstrated high cellular retention of 90–95 % of the radioactivity with a washout half-life greater than 500 h. By contrast, although the 131I labeled J591 bound effectively to PSMA-expressing LnCaP cells in suspension, the 131I label was observed to wash out of the cell as the iodine had likely labeled tyrosine moieties within the immunoglobulin. Hence, subsequent evaluation was limited to the radiometals 177Lu and 90Y (Smith-Jones et al. 2000). Significant anti-tumor responses were observed in LnCaP xenograft tumor bearing nude mice and a dose response relationship was observed (Fig. 5). Higher cumulative doses of either 90Y or 177Lu were possible using fractionated dosing (multiple submaximal tolerated dose (MTD)) rather than a single MTD dose. Median survival of the animals improved threefold for fractionated 90Y-muJ591 therapy versus control (150 vs. 52 days). With fractionated doses of 177Lu-muJ591, >80 % of the mice were cured (Smith-Jones et al. 2003).
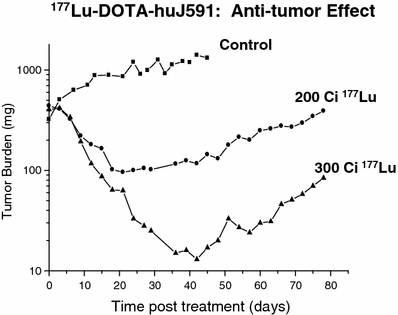
Fig. 5
Effect of incremental activity of Lu-177 vs control on tumor mass (mg) in-planted into nude mice. A greater anti-tumor effect is demonstrated at 300 microCuries vs 200 microCuries. The tumor continued to enlarge in the control mice
3.2 Preclinical Studies with Radiolabeled Heterodimer PSMA Antagonists
Following the identification of a series of urea-amino acid based molecules that inhibited the glutamate carboxypeptidase enzymatic activity of PSMA, Babbich, Pomper, and co-workers evaluated several of these molecules that were essentially lysine-urea-lysine or glutamate-urea-lysine analogs (Hillier et al. 2009). One of these molecules, identified as MIP 1095 had a Ki of 0.24 + 0.14 nmol/L indicating high binding affinity. When labeled with 123I, the affinity for human prostate cancer LnCaP cells was 0.81 + 0.39 nmol/L. Similarly, high affinity for PSMA was demonstrated for other similar molecules. This binding was also specific for PSMA as there was no binding to prostate cancer cells that did not express PSMA. This high affinity, selective targeting, was subsequently demonstrated in nude mice bearing LnCaP xenografts (Fig. 3).
4 Clinical Studies
4.1 Radioimmunotherapy
Following “humanization” of the murine antibody by removal of a substantial portion of the murine immunoglobulin and substituting nonspecific human gamma globulin for most of the molecule but retaining the immuno-recognition portion, initial clinical studies were performed to determine the safety and effect on biodistribution of incremental amounts of the humanized antibody using 111In DOTA-J591. Repetitive dosing was well tolerated up to 500 mg without the development of a human anti-humanized (de-immunized) antibody (HAHA) response or evidence of dose limiting toxicity (DLT); the maximum tolerated dose (MTD) was not reached (Bander et al. 2000). Monoclonal antibody targeting to normal tissues was not observed but significant uptake of the radiolabel was seen in the liver, typical of radiometal labeled immunoglobulin biodistribution patterns (Fig. 6). The liver activity diminishes with increasing dose of antibody but a considerable fraction of the injected dose appears in the liver. Increasing the amount of antibody infused results in longer plasma clearance times (Bander et al. 2001; Vallabhajosula et al. 2005a). These data demonstrated that relatively low amounts of total immunoglobulin, in the order of 20 mg, were sufficient to minimize hepatic uptake and maximize plasma circulation time. Subsequently, a total of 20 mg of the J591 antibody in all clinical studies; that is after labeling at a specified specific activity demonstrated not to adversely influence immuno reactivity, the individual patient doses were supplemented with unlabeled humanized J591, so that the preparation to be administered contained a total of 20 mg/m2 of immunoglobulin.
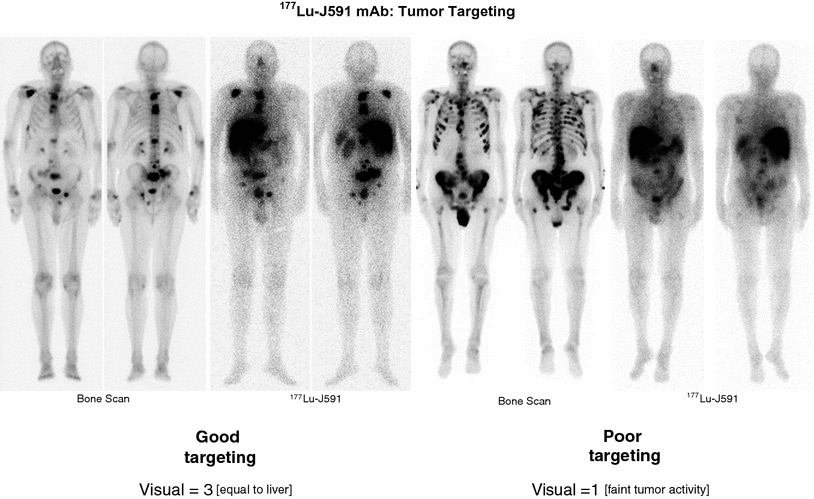
Fig. 6
 (Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
(Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
 Thyroid Carcinoma
Thyroid Carcinoma
 of Tumors: Pretargeting with Bispecific Antibodies
of Tumors: Pretargeting with Bispecific Antibodies
 Imaging for Radiation Therapy Planning
Imaging for Radiation Therapy Planning
 Radiopharmaceuticals for PET Dosimetry Before, During, and After Endoradiotherapy
Radiopharmaceuticals for PET Dosimetry Before, During, and After Endoradiotherapy
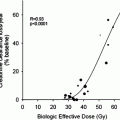 Use of Dosimetry in the Planning of Patient Therapy
Use of Dosimetry in the Planning of Patient Therapy




Bone scintigraphy and corresponding gamma scintigraphy of patients receiving Lu-177 J591 demonstrating (left) good targeting and (right) poor targeting. In patients with scores less than 3, only 1 of 19 patients had a PSA decline greater than 30 % whereas at better targeting (>4), 5 of 20 patients had a declinegreater than 30 % and 10 of 20 had a 10–30 % decline. (See text and references for details)
Related posts:
(Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
Thyroid Carcinoma
of Tumors: Pretargeting with Bispecific Antibodies
Imaging for Radiation Therapy Planning
Radiopharmaceuticals for PET Dosimetry Before, During, and After Endoradiotherapy
Use of Dosimetry in the Planning of Patient Therapy
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
