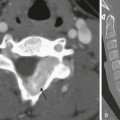
Chapter 116 Overview and Pathophysiology: Nephrogenesis is completed by the 36th gestational week. A focus of fetal metanephric blastema or embryonal renal tissue that persists beyond 36 weeks gestation is called a nephrogenic rest. Multiple foci or diffuse nephrogenic rests are termed nephroblastomatosis. Nephrogenic rests are identified in about 1% of neonatal autopsies but usually are no longer found after 4 months of age. Malignant transformation of the fetal metanephric blastema may occur, along with development of a Wilms tumor (nephroblastoma), or nephroblastomatosis may resolve spontaneously.1–3 Nephrogenic rests can occur anywhere in the kidney, depending on when nephrogenesis is interrupted. These rests may be located in the renal lobe (intralobar) or in the cortex that envelops the renal lobe (perilobar). The two types of nephrogenic rests have different appearances, malignant potential, and associated genetic abnormalities. Nephrogenic rests also are classified according to histologic features of development; hyperplastic and neoplastic rests are thought to be active and to have malignant potential, whereas dormant or sclerosing nephrogenic rests are considered inactive.1–4 Intralobar nephrogenic rests are less common than perilobar nephrogenic rests and are more likely to degenerate into a Wilms tumor. Intralobar nephrogenic rests tend to be few and are located randomly in the renal lobe; they are seen in patients with sporadic aniridia, Drash syndrome (i.e., male pseudohermaphrodism and nephritis), and WAGR syndrome (i.e., Wilms tumor, aniridia, genital anomalies, and mental retardation). Patients with sporadic aniridia have a 30% to 40% risk of having a Wilms tumor develop, which represents the greatest likelihood of all of the genetic and syndromic abnormalities associated with nephroblastomatosis.1–4 Perilobar nephrogenic rests are multiple and are located at the corticomedullary junction or in the cortex. Also called diffuse perilobar nephrogenic rests or diffuse perilobar nephroblastomatosis, they are found in persons with hemihypertrophy and in persons with Beckwith-Wiedemann syndrome (i.e., macroglossia, macrosomia, and omphalocele), Perlman syndrome (i.e., fetal gigantism and multiple congenital anomalies), and trisomy 18 syndrome. Patients with hemihypertrophy and Beckwith-Wiedemann syndrome have about a 5% risk of having a Wilms tumor develop.1,3–5 Imaging: Microscopic nephrogenic rests cannot be identified radiologically. Diffuse perilobar nephroblastomatosis and multifocal nephroblastomatosis can be evaluated through ultrasonography, computed tomography (CT), and magnetic resonance imaging (MRI). In diffuse perilobar nephroblastomatosis, the affected kidney may be enlarged. On sonography, corticomedullary differentiation is absent. Regions of nephroblastomatosis may be hypoechoic or isoechoic with respect to normal renal cortex (Fig. 116-1). Multifocal nephroblastomatosis is more difficult to identify on sonography.2,3,6 Figure 116-1 Nephroblastomatosis. CT is more sensitive than ultrasound for the evaluation of nephroblastomatosis. On contrast-enhanced CT, areas of nephroblastomatosis are well defined because they enhance to a lesser extent than does normal renal cortex (Fig. 116-2). Bulky masses of nephroblastomatosis may distort the pelvicalyceal system. Involvement may be symmetric or asymmetric (e-Fig. 116-3). In diffuse perilobar nephroblastomatosis, a thick rind of lower attenuation tissue encases normally enhancing but architecturally distorted parenchyma. In persons with multifocal nephroblastomatosis, multiple round masses of low attenuation are present. Flat or plaquelike areas of involvement may be difficult to identify on CT.2,3,6 Figure 116-2 Diffuse perilobar nephroblastomatosis with symmetric involvement. e-Figure 116-3 Diffuse perilobar nephroblastomatosis with asymmetric involvement. On MRI, nephroblastomatosis cannot be distinguished from normal renal parenchyma on T1-weighted sequences and is isointense or hyperintense on T2-weighted sequences. Nephroblastomatosis appears hypointense relative to normal renal parenchyma after administration of contrast material on T1-weighted sequences, which are the most sensitive for detection. Active areas of nephroblastomatosis typically are hyperintense on T2-weighted sequences, and inactive areas are hypointense. The signal intensity of nephroblastomatosis is homogeneous, whereas foci of Wilms tumor tend to be heterogeneous.2,3,6–8 Treatment and Follow-up: Although the subject is somewhat controversial, presently no specific treatment is advocated for nephrogenic rests/nephroblastomatosis. Close radiologic follow-up is recommended for children with genetic abnormalities or syndromes associated with nephroblastomatosis. Patients should be monitored to detect Wilms tumor development, because the prognosis for Wilms tumor is best with small lesions. Children with hemihypertrophy or Beckwith-Wiedemann syndrome are at risk for the development of other embryonal tumors, such as hepatoblastoma and adrenal cell carcinoma. No large studies have been performed to establish the optimal screening interval for Wilms tumor surveillance; however, large tumors with metastases that develop within a 6-month period are reported. A baseline CT of nephroblastomatosis-related genetic abnormalities or syndromes at 6 months of age (or at diagnosis if the patient is older than 6 months), followed by ultrasound examinations every 3 to 4 months until the child is 8 years of age, is recommended on the basis of results from the National Wilms Tumor Studies. An area of nephroblastomatosis that grows larger and rounder is suspicious for malignant degeneration.2,3,9–13 Overview, Pathophysiology, and Staging: Wilms tumor is the most common abdominal malignancy of childhood and accounts for 87% of renal masses. Its peak incidence is at 3 to 4 years of age (80% occur in children <5 years), but it has been described in the fetus, neonate, teenager, and adult. Clinical presentation includes a palpable mass, abdominal pain, hematuria, and occasional hypertension (from tumor renin production). As discussed in the Nephroblastomatosis Complex section of this chapter, certain syndromes and genetic abnormalities predispose to development of a Wilms tumor (e-Fig. 116-4). Two loci on chromosome 11 have been implicated in the genesis of Wilms tumors: 11p13 (WT1 gene—WAGR or Drash syndrome) and 11p15 (WT2 gene—Beckwith-Wiedemann syndrome or hemihypertrophy). Bilateral Wilms tumors occur almost exclusively in patients with nephroblastomatosis. Most Wilms tumors arise from the renal parenchyma; however, extrarenal Wilms tumors rarely may develop in the abdomen or at distant sites.1,14–17 e-Figure 116-4 Diffuse perilobar nephroblastomatosis and a right Wilms tumor. In the United States, tumor stage (Box 116-1) is determined operatively, and the grade is established on pathologic examination. The classic triphasic Wilms tumor arises from mesodermal precursors of the renal parenchyma (metanephros) and contains blastemal, stromal, and epithelial elements. The “teratoid Wilms tumor” contains tissue not normally found in the kidney (e.g., bone, cartilage, and muscle). Tumors with a favorable histology do not contain any anaplastic changes. The prognosis for tumors with a favorable histology is excellent, even for those at higher stages. In Europe, the staging system is based completely on radiologic findings. Tumors are classified on the basis of their imaging appearance, and chemotherapy is given before definitive surgery is performed. Tumors with extension into the inferior vena cava or invasion through the renal capsule are easier to resect after tumor shrinkage produced by chemotherapy.1,2,18 Imaging: Radiologic evaluation of Wilms tumor is focused on identifying the site(s) of involvement, extension, and metastases to assist in surgical planning. Preoperative imaging may include conventional chest radiography, abdominal and pelvic sonography, and thoracic, abdominal, and pelvic CT, MRI, and potentially fluorine deoxyglucose positron emission tomography (FDG-PET) fused with CT (PET-CT).1,2,19,20 On sonography, a Wilms tumor is an intrarenal mass of heterogeneous echogenicity. Some Wilms tumors may contain cystic components, portions of obstructed and entrapped pelvicalyceal systems, or hemorrhagic and necrotic tumor. Extension into the inferior vena cava and the right atrium are characteristic routes of tumor growth and can be well visualized on ultrasound (e-Fig. 116-5), CT (Fig. 116-6), and MRI. The tumor typically forms a pseudocapsule but may invade the renal capsule, seed the peritoneal space, or grow directly into the mesentery and omentum. Hepatic metastases also are possible. The contralateral kidney may contain a smaller Wilms tumor or nephroblastomatosis. Synchronous or metachronous bilateral Wilms tumor may occur in up to 10% of patients.1,2,6 Figure 116-6 A Wilms tumor extending into the inferior vena cava and right atrium. e-Figure 116-5 A Wilms tumor extending into the inferior vena cava and the right atrium (RA). On CT, Wilms tumor is generally spherical and intrarenal. It may contain small amounts of fat or fine calcification (Fig. 116-7 and e-Fig. 116-8) because the metanephric blastema cell is a pluripotential embryonal cell. Dystrophic calcifications are seen in about 9% of Wilms tumors. The tumor enhances to a lesser extent than does renal parenchyma. In patients who have been screened ultrasonographically because of genetic or syndromic conditions, the tumor is usually smaller than 4 cm in diameter. Children who are evaluated because of physical examination abnormalities generally have tumors larger than 10 cm in diameter. CT and conventional radiographs of the chest are used to identify pulmonary metastases.1,2,6,21,22 Figure 116-7 A Wilms tumor containing fat in a horseshoe kidney. e-Figure 116-8 A Wilms tumor containing calcification. On MRI, a Wilms tumor is isointense with respect to normal renal parenchyma on T1-weighted sequences and hyperintense with respect to normal renal parenchyma on T2-weighted sequences. After administration of contrast material, a Wilms tumor is hypointense relative to normal renal parenchyma and has inhomogeneous signal intensity (Fig. 116-9). An effectively treated Wilms tumor may be hypointense on T2-weighted sequences.1,2,7,8,23 Figure 116-9 A Wilms tumor. FDG-PET fused with CT is a rapidly developing modality for imaging accelerated metabolism in malignant tissue. FDG-PET has better accuracy than MRI and bone scanning for staging. Wilms tumors are presumed to be FDG-avid, but the role of FDG in Wilms evaluation is as yet unclear. FDG-PET can help with a targeted biopsy of a viable Wilms tumor and biologically aggressive elements (e.g., anaplastic Wilms). Sensitivity for lung metastases is dependent on nodule size and respiratory motion. Treatment response of the primary tumor can be monitored. Potential benefits of FDG-PET will require ongoing investigation.2 Treatment: Surgery for a Wilms tumor typically begins with a complete abdominal exploration before attention is given to the kidneys. The unaffected kidney initially is visualized and palpated to assess for masses or areas of superficial nephroblastomatosis before en bloc resection of the affected kidney is performed. An excisional biopsy of lung nodules is performed to ensure that the stage of the tumor is correctly assigned. Patients with bilateral disease undergo staging surgery as well. Results of the surgical staging and the histologic examination determine the selection of therapy. In patients with bilateral disease, treatment is provided according to the tumor histology of the higher stage side, along with later nephron-sparing surgery. When the tumor is very large or when the tumor extends into the right atrium via the inferior vena cava, the surgeon may defer surgery until several courses of chemotherapy have reduced the tumor size or extension. Substantial progress in therapy for persons with a Wilms tumor over recent decades has resulted in a greater than 90% long-term survival for localized disease and greater than 70% survival for metastatic disease.1,2,24–30 Overview: Clear cell sarcoma of the kidney accounts for approximately 5% of primary renal tumors in childhood and was considered a sarcomatous subtype of Wilms tumor before 1978. It was reclassified as a separate entity from Wilms tumor because of the tumor’s histologically and biologically unique features. No familial or syndrome association has been identified. Clear cell sarcoma occurs in an age group similar to that affected by Wilms tumor (1 to 4 years of age), and has a male predominance. Immunohistochemical staining has shown no characteristic marker pattern, but negativity to the WT1 gene is important. Imaging and Treatment: Clear cell sarcoma imaging features are not distinct from those of the Wilms tumor (Fig. 116-10). Clear cell sarcoma has a predilection for bone metastases (formerly known as the bone metastasizing renal tumor of childhood) that may occur at presentation or upon relapse. A pathologic diagnosis of clear cell sarcoma necessitates an evaluation of the skeletal system. Bone scintigraphy or a conventional radiographic skeletal survey may be used. However, metastases to lymph nodes, lung, and liver still are seen more frequently than bone metastases. Because of the aggressive behavior of clear cell sarcoma, it is associated with a higher rate or relapse and mortality than is Wilms tumor, with a reported long-term survival of 60% to 70%. Treatment consists of nephrectomy and aggressive chemotherapy.1,31–38
Renal Neoplasms
Nephroblastomatosis Complex
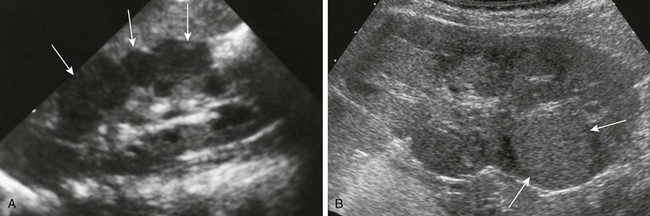
A, Diffuse perilobar form. A longitudinal sonogram shows an enlarged right kidney with lobulated contour and hypoechoic cortical masses (arrows). B, Focal intralobar form. A longitudinal sonogram shows a lower pole mass (arrows) that is isoechoic with renal cortex.
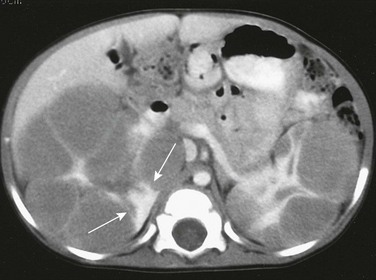
On a contrast-enhanced computed tomography scan, the kidneys are enlarged and multiple, round, peripheral masses of low attenuation are present. Architectural distortion at this level is pronounced; in the medial portion of the right kidney, only a peripheral area of normally enhancing kidney (arrows) is present.
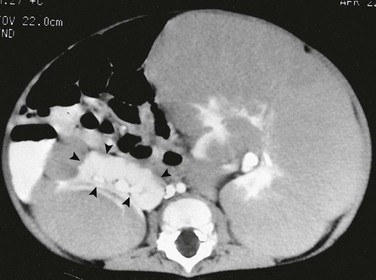
On a contrast-enhanced computed tomography scan, the left kidney is much larger than the right. The architecture of the kidneys is distorted. A portion (arrowheads) of the normally enhancing right kidney consists of parenchyma and calyces.
Wilms Tumor
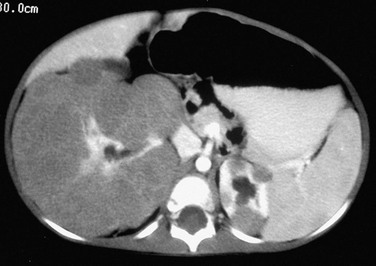
A contrast-enhanced computed tomography scan shows round, peripheral masses that enhance to a lesser extent than normal renal parenchyma in the upper pole of the left kidney. The right kidney has a thick rind of lobulated, hypoattenuating tissue without a discreet mass. A biopsy showed Wilms tumor on the right, and a nephrectomy was performed.
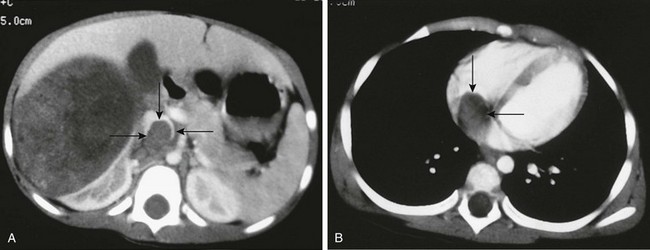
A, A contrast-enhanced computed tomography scan reveals a large, round, heterogeneous mass in the right kidney that enhances to a lesser extent than normal renal parenchyma. Contrast material opacifies the lateral and anterior margins of the inferior vena cava that contain hypoattenuating tumor (arrows). B, The tumor (arrows) extends into the right atrium.
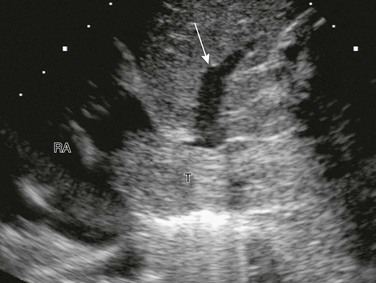
A longitudinal sonogram shows tumor thrombus (T) expanding the inferior vena cava and extending into the RA. The tumor partially occludes drainage from one of the hepatic veins (arrow).
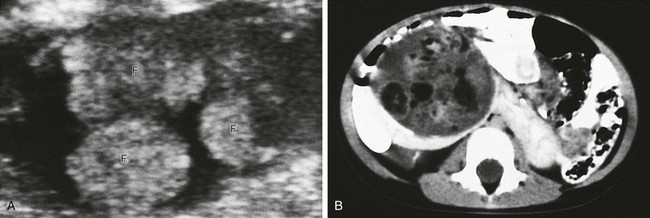
A, An axial sonogram of part of the Wilms tumor reveals hyperechoic lobules containing fat (F). B, A contrast-enhanced computed tomography scan shows areas of fat within the mass. The isthmus of the renal parenchyma extends across the midline.
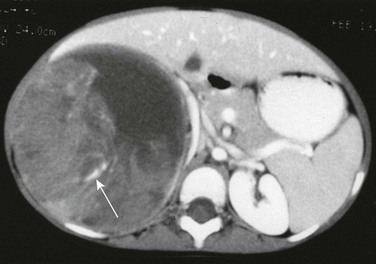
A contrast-enhanced computed tomography scan reveals fluid-attenuating material anteromedially, most likely hemorrhage, within the large heterogeneous mass. Linear calcification is present posteriorly (arrow).
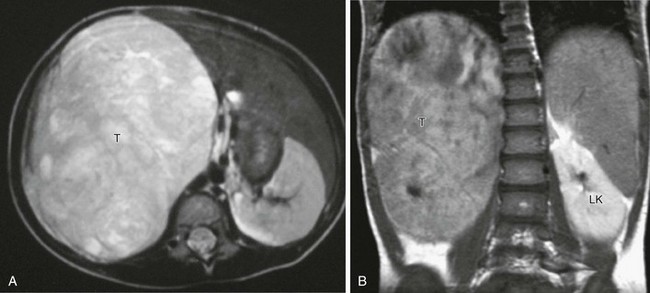
A, Axial T2-weighted magnetic resonance imaging (MRI) shows a large tumor (T) replacing the right kidney, which is hyperintense to other soft tissue structures. B, Coronal T1-weighted MRI after gadolinium in the same patient shows that the large tumor (T) is hypointense relative to the normally enhancing left kidney (LK).
Clear Cell Sarcoma of the Kidney
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree