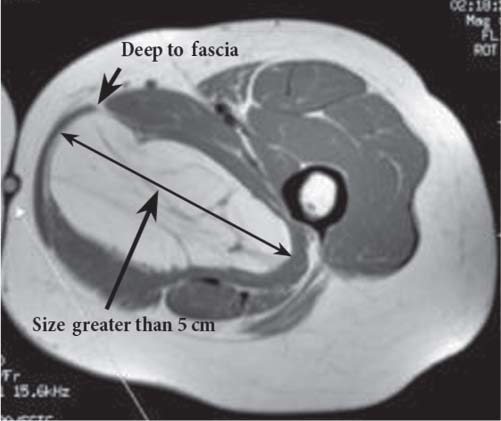
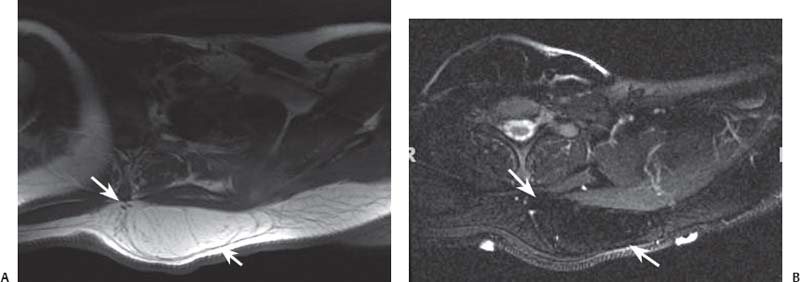
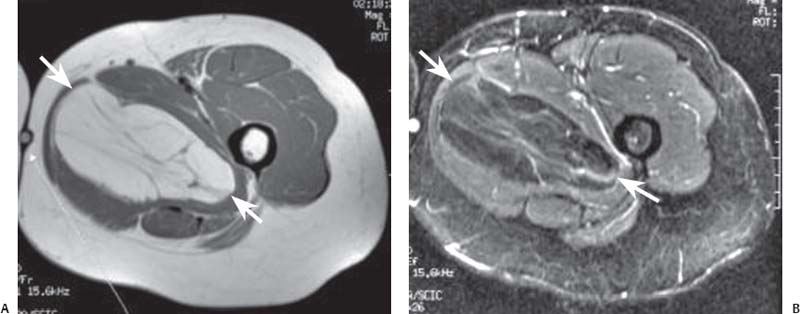
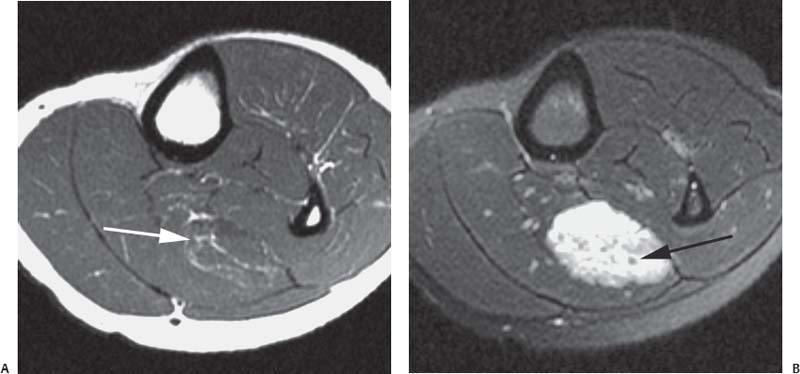
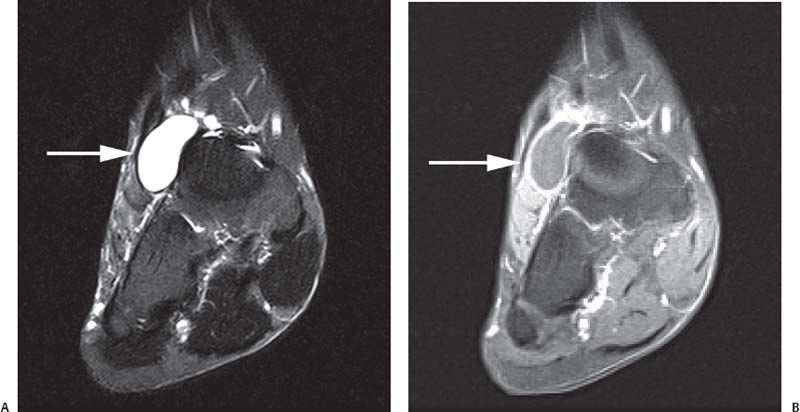
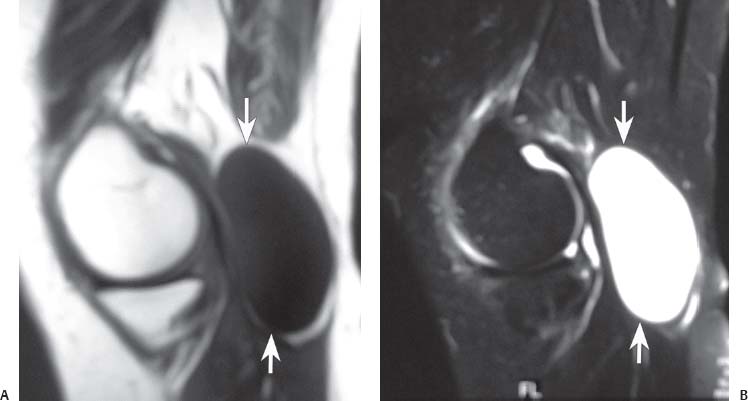
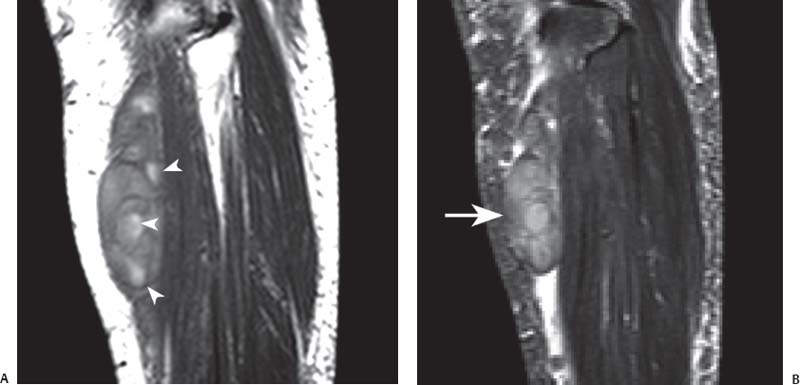
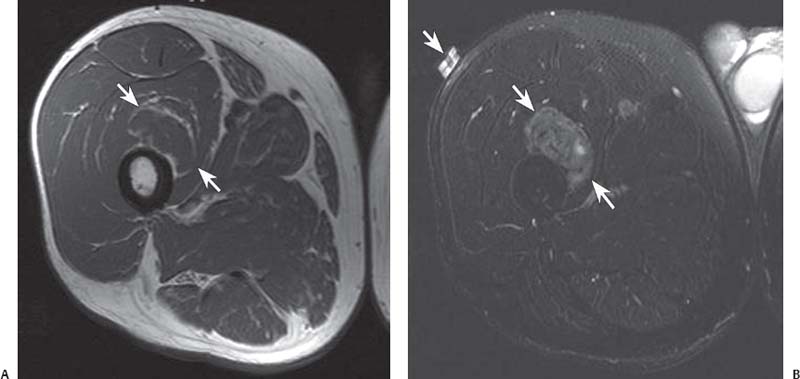
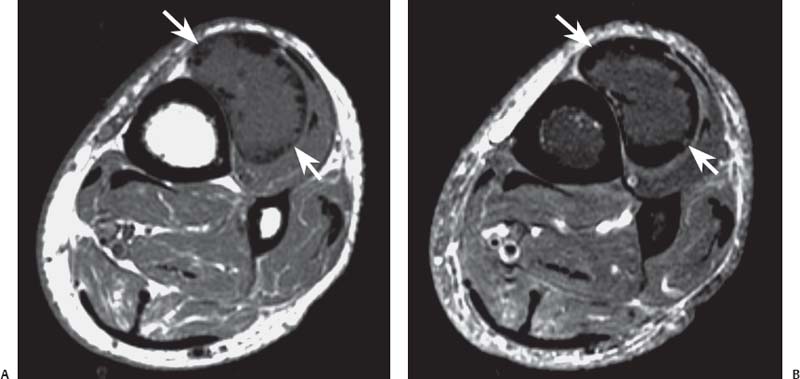
15 Soft-Tissue and Bone Tumors The management of soft-tissue masses presents an interesting quandary for physicians that has important implications. Although it has been suggested that 1 of 100 soft-tissue lesions seen by a physician is malignant, the precise overall number is unknown.1 The incidence of soft-tissue sarcomas has been estimated to be approximately 8,100 cases per year in the United States.2 In most cases, soft-tissue masses are benign. Indeed, many of these soft-tissue lesions have no potential for metastasis or local invasion and can simply be observed. The danger in simple observation occurs when the physician observes a lesion without having a firm diagnosis; this situation can lead to errors in management with resultant poor outcomes, including local invasion of neurovascular structures and metastatic disease. Similarly, excision of a lesion without a definitive diagnosis can also result in catastrophic outcomes. With an incorrect diagnosis, excision of a malignant lesion can lead to the contamination of unaffected tissues, recurrence, and, in some cases, eventual amputation of an affected limb. For many years, the use of tissue biopsy was the only means of obtaining a definitive diagnosis. Today, the increased use of MRI has substantially improved the diagnosis and management of soft-tissue tumors. MRI provides excellent soft-tissue resolution and allows the physician to differentiate various soft-tissue types based on imaging characteristics via the use of various pulse sequences,3,4 a feature not afforded by other imaging modalities such as conventional radiographs and CT. The excellent spatial resolution provided by MRI also provides sharp delineation of soft-tissue boundaries and highlights the boundaries between the soft-tissue tumor and the adjacent normal tissues. This information can help guide the determination of the diagnosis of a soft-tissue lesion, which can obviate tissue biopsy. Another advantage of MRI relates to its use in preoperative planning. With the guidance of multiplanar imaging provided by MRI, neurovascular structures can be avoided during the approach to the lesion, eliminating or decreasing the potential for contamination during planned biopsies or providing a means to ensure adequate margins when resection is planned. With all of its inherent capabilities, MRI has become a powerful tool in the diagnosis and management of soft-tissue tumors. In addition to its value in diagnosing soft-tissue tumors, the diagnosis of bone tumors has been enhanced by the use of MRI in conjunction with conventional radiographs and CT. Although conventional radiographs often provide enough information to make a diagnosis of a given bone tumor, MRI can provide additional details that will help guide the management of the lesion. MRI also allows for the visualization of pathology that cannot be seen on conventional radio-graphs, such as fluid–fluid levels or the degree of invasion of the adjacent soft-tissue structures. A lesion’s tissue composition also can be identified more easily with MRI than with conventional radiographs. This information can affect the surgeon’s management decisions, such as the choice of neoadjuvant or postoperative chemotherapy. The clinician must properly diagnose the soft-tissue lesion before planning any type of treatment, including simple observation. Understanding this point and the consequences of misdiagnosis, the surgeon must take a methodical, systematic approach to the diagnosis of these lesions. Therefore, a diagnostic and treatment algorithm directed toward soft-tissue and bone lesions depends on information from the history, physical examination, conventional radiographs, CT scans, and MRI studies. This chapter provides such an algorithm based on the concept of determinate versus indeterminate soft-tissue tumors, reviews common soft-tissue and bone lesions, and describes them and their characteristic MRI findings. As with any medical condition, the evaluation of a soft-tissue tumor begins with the history and physical examination. Although they are essential parts of the diagnostic process, these two items often do not provide a definitive diagnosis for soft-tissue lesions. Most soft-tissue tumors are slow-growing lesions. A history of trauma, although suggestive of a hematoma or heterotopic ossification, does not ensure that the lesion is a benign, posttraumatic process. The trauma may merely alert the patient to a soft-tissue tumor that had previously been present in the area. In addition, it is important to note that a history of pain is not a reliable indicator of the benign or malignant nature of a lesion and that only half of all patients with a malignant soft-tissue mass complain of pain.5 Systemic symptoms, such as fever, malaise, chills, or night sweats, are associated with the presence of malignant lesions, but the absence of systemic complaints does not necessarily indicate that the lesion is benign. Ultimately, the history may assist the physician in diagnosis, but the lack of consistency in soft-tissue tumor symptomatology often means that the history alone is seldom beneficial in definitively diagnosing soft-tissue lesions. Fig. 15.1 An axial, postgadolinium, T1-weighted image of the left proximal thigh showing a lesion that displays two of the common signs of a malignant soft-tissue lesion: size >5 cm and location deep to the fascia. Fig. 15.2 Lipoma of the left upper back. (A) An axial T1-weighted image shows a high signal intensity lesion (arrows) with the same intensity of surrounding fat. The lesion itself is homogeneous and superficial. (B) An axial STIR image shows suppression of the signal within the lesion (arrows). Fiduciary markers on the skin delineate the lesion. Similarly, the physical examination is frequently nonspecific in nature and often cannot be used to determine whether a lesion is benign or malignant. It is extremely helpful in some cases (e.g., periarticular ganglion cysts), but, for most soft-tissue masses, physical examination does not provide a definitive answer. For example, although a size of >5 cm, a location beneath the fascia, and a feeling of firm or matted material are associated more with malignant than with benign lesions, none of these criteria is pathognomonic for malignancy (Fig. 15.1).6 At the same time, malignant lesions can present as small or nongrowing lesions. Sarcomas are typically large, but to characterize a small lesion as benign without having a definitive diagnosis is a mistake. Neurologic deficit can result from malignant or benign lesions. It has been reported that certain lesions have characteristic appearances on MRI and other imaging modalities, which allows the physician to diagnose the lesion with a high degree of confidence.7 Lesions in this category are termed determinate. Lesions that cannot be diagnosed with certainty without biopsy are termed indeterminate. With determinate lesions, that is, those with a distinctive appearance on a radiograph or MRI or a very characteristic physical examination, the physician feels comfortable making a diagnosis without a tissue sample and can make the appropriate decision about treatment without gathering additional information. It is important to remember that, as with all tumors, good communication between the treating physician and physicians from other disciplines is essential to making the proper diagnosis. Discussions with an experienced musculoskeletal radiologist can help guide or confirm the physician’s diagnosis, as can discussions with a pathologist, especially one with a special interest or advanced training in the evaluation of soft-tissue and bone tumors. A multidisciplinary approach reduces the risk of diagnostic errors.7 Each physician will develop his or her own level of comfort in classifying such lesions. Lipomas, the most common of all soft-tissue tumors, are composed of mature fatty tissue. Although typically asymptomatic, these lesions can cause pain and neurologic symptoms by compressing neurovascular structures. Lesions that cause such symptoms usually lie deep to the fascia. Because lipomas consist of mature fatty tissue, the signal intensity of a lipoma on MRI exactly matches the intensity of subcutaneous fat. On routine SE pulse sequences, this appearance translates to high signal intensity on T1-weighted images and moderate to high intensity on T2-weighted images. When lipomas are suspected, the use of a fat-suppression technique and STIR images can confirm the lipomatous nature of the lesion by suppressing the high signal intensity related to the adipose tissue.7 The tissue seen in the lipoma should match the signal characteristics of subcutaneous fat on all pulse sequences, including fat-suppressed and STIR images (Fig. 15.2). Fibrous septations may appear as hypointense thin lines that may or may not enhance with contrast.8 Lipomas, especially those in superficial or subcutaneous locations, are well-demarcated lesions that do not invade surrounding structures. However, deep lipomas may surround vascular and neural structures. Lipomatous variants that contain other mesenchymal elements, such as fibrous or myxoid tissue, differ from the typical lipoma. The physician must evaluate atypical lipomas carefully. Differentiating them from low-grade liposarcomas without a tissue sample may be dificult, if not impossible. Areas of heterogeneity on MRI should alert the physician to possible malignancy. Broad septations or septations with nodules are characteristics of well-differentiated liposarcomas (Fig. 15.3). Hemangiomas are relatively common benign soft-tissue tumors composed of benign blood vessel elements. Estimates of female-to-male predominance range as high as 3:1,9 although other investigators have not shown that same distribution.7 These lesions occur most commonly in children, and cutaneous manifestations of the lesion usually spontaneously involute in the first decade of life. Other manifestations include the following: • Cavernous subtype lesions • Venous lesions • Arteriovenous lesions • Mixed-type lesions Hemangiomas that prove more diffcult to diagnose are those that lie deep in the soft tissues. Superficial blood vessel tumors can have a spongy or fluctuant quality, and, on occasion, such lesions fill or swell when the limb is placed in a dependent position. In contrast, lesions that lie deep to the fascia may not have any unique findings on physical examination and can only be palpated; these lesions can increase in size during pregnancy.10 Approximately half of these lesions cause pain after exertion, which may relate to a vascular steal phenomenon and the resultant tissue ischemia when the lesion absorbs blood flow or results in retrograde flow.11 Fig. 15.3 Atypical lipoma in the left thigh. (A) An axial T1-weighted image shows a large, high signal intensity lesion (arrows) with multiple septations, suggestive of an atypical lipoma. (B) An axial STIR image shows suppression of most of the signal within the lesion (arrows) but also shows multiple fibrous septations with a complex, heterogeneous composition. Because of its large size and heterogeneous appearance, this lipoma was biopsied. Phleboliths may be seen on conventional radiographs in approximately 50% of patients with hemangiomas.11 Phleboliths appear as small, round, mineralized soft-tissue densities that have a lucent center. A nondescript soft-tissue mass may be seen. The MRI is often diagnostic. A hemangioma is a heterogeneous mass that can contain varying degrees of thrombus, hemosiderin, vessel formation, fibrosis, and fat. The amount of fat can be substantial. With T1-weighted SE imaging, the areas corresponding to adipose tissue show high signal. The lipomatous portion usually involves the periphery of the lesion. Blood-filled cavernous or vessel components of the lesion also appear bright with T2-weighted and STIR imaging, and they enhance on postgadolinium T1-weighted images (Fig. 15.4). Depending on the nature of the vessel formation, a “serpentine” figure may be apparent; septations and lobules are easily recognized. If there is rapid flow within the lesion, flow voids or focal regions of low signal on T2-weighted or STIR images are seen.12 On ultrasound, the lesions appear echogenic, and color-flow Doppler often shows obvious flow within the lesion.13 Another common determinate lesion, the ganglion cyst, arises from periarticular tissues and tendon sheaths. This lesion is composed of viscous mucinous fluid contained by a thick fibrous shell, but debate still exists over its etiology. Some believe that repeated stress causes mucoid degeneration,14,15 whereas others hypothesize that lining cell hyperplasia with production of a hyaluronic acid-rich substance causes degeneration into a cystic lesion.15,16 The most common locations for a ganglion cyst include the dorsal and volar aspects of the wrist. Because the lesion typically occurs in this location and has a characteristic appearance, the physician often can make the diagnosis without additional imaging. Other common locations include the following: • Foot and ankle • Tendon sheaths • Labra • Joint capsules Fig. 15.4 Hemangioma. Axial T1-weighted (A) and axial fat-suppressed T2-weighted (B) images of the right calf show a mass (arrow) in the medial head of the gastrocnemius muscle. The mass is minimally high signal on the T1-weighted image and heterogeneous and serpentine high signal on the STIR image. (From Papp DF, Khanna AJ, McCarthy EF, Carrino JA, Farber AJ, Frassica FJ. Magnetic resonance imaging of soft tissue tumors: determinate and indeterminate lesions. J Bone Joint Surg Am 2007;89(suppl 3):103-115. Reprinted by permission.) The ganglion cysts often do not communicate with a joint. When the lesion is located in other areas, additional imaging often is needed. On conventional radiographs, osseous erosion is occasionally seen as a result of pressure erosion. MRI delineates the cyst as a smooth, round, or ovular well-circumscribed structure that may have septations. These septations, or the peripheral rim of the lesion, may enhance with gadolinium, but the center of the lesion should not enhance with contrast. T1-weighted imaging shows decreased signal intensity, and lesions appear bright on T2-weighted or STIR imaging, with signal intensity similar to that of water (Fig. 15.5).4,12,17–19 Although a ganglion cyst rarely arises inside of a joint and does not communicate with the joint itself, the synovial (Baker) cyst is contiguous with the joint space. Classically described by Baker,20 the lesion arises from synovial fluid by pushing its way from the joint into a communicating bursa (often under the medial head of the gastrocnemius at the knee) or by causing herniation of the synovial membrane itself. The lesion arises in the popliteal fossa and may cause posterior knee pain. Occasionally, these cysts can cause complications, such as the following21,22: • Cyst leakage • Thrombophlebitis • Compartment syndrome • Lower limb claudication Meniscal tears have been described as the most common etiology of these lesions, although the cyst can form from other intraarticular processes, such as degenerative arthritis or ACL injury. The prevalence of this entity in adults ranges from 5% to 20%.23,24 Approximately half of patients with osteoarthritis have a Baker cyst.25 Excision of the cyst may not provide relief because the cysts commonly recur. Treatment of the underlying condition often results in resolution of the cyst. Radiographs often show osteoarthritis, which (as described above) can cause the development of the disease. They do not show the process itself. Although ultrasound is another modality that can show synovial cyst formation, MRI is the standard for diagnosis because the underlying pathology, such as ACL or meniscal tears, also can be evaluated with this modality. MRI shows a well-circumscribed mass in the posterior fossa of the knee. T1-weighted and T2-weighted images show fluid with the same intensity as that of joint fluid (dark on T1-weighted images and bright on T2-weighted images) (Fig. 15.6). The addition of gadolinium contrast shows enhancement of only the rim. Fig. 15.5 Ganglion cyst. Coronal STIR (A) and coronal contrast-enhanced, fat-suppressed T1-weighted (B) images show the typical appearance of a ganglion cyst (arrow) along the dorsum of the midfoot. The lesion is round, well-circumscribed, and hyperintense compared with muscle on the STIR image with a thin rim of enhancement, which is compatible with the fluid-filled, cystic nature of a ganglion cyst. (From Papp DF, Khanna AJ, McCarthy EF, Carrino JA, Farber AJ, Frassica FJ. Magnetic resonance imaging of soft tissue tumors: determinate and indeterminate lesions. J Bone Joint Surg Am 2007;89(suppl 3):103-115. Reprinted by permission.) Fig. 15.6 Synovial cyst. (A) A sagittal T1-weighted image shows a large, low signal intensity mass (arrows) posterior to the knee joint. (B) A sagittal T2-weighted image shows the same lesion (arrows) to be of high signal intensity and well circumscribed, compatible with a cyst (in this case, a Baker cyst). The differentiation of a hematoma from hemorrhage within a sarcoma can be difficult. Although not definitive, a history of trauma favors the diagnosis of a hematoma. A history of bruising or ecchymosis noted on physical examination may help with the diagnosis. Most patients with a hematoma have a history of trauma or surgical intervention. The natural history of hematomas follows one of three pathways: • Spontaneous involution • Development of peripheral calcification and progression to myositis ossificans (see below) • Chronic expansion of the hematoma The clinical picture for the last presentation differs somewhat from that of the other two, as described by Reid et al.26 Typically, a patient presents with a slow-growing soft-tissue mass that does not involute within a month of the initial injury. Some clinicians believe that the hemosiderin breakdown products do not allow the lesion to heal completely and that the persistent irritation caused by these products maintains patent capillary bleeding into the hematoma.26–28 Systemic anticoagulation has been described as a cause of the formation of these lesions.28 Often painless, these lesions can produce neurologic deficits via neurovascular compression. Radiographs may show pressure erosion of the surrounding bones.27 The MRI findings are characteristic. T1-weighted images usually show a well-defined mass. Centrally, the lesion is heterogeneous, with bright foci that correspond with areas of new or continuing hemorrhage. T2-weighted images also show heterogeneity, with areas of high and low signal intensity corresponding to granulation tissue and hemosiderin deposition, respectively. The presence of a low signal intensity pseudocapsule completes the picture (Fig. 15.7). Gradient-echo images may help isolate hemosiderin in the lesion.27,28 When the lesion does not show any enhancement with gadolinium contrast, the diagnosis is most likely a hematoma. Enhancement of the pseudocapsule has been described with gadolinium, but it is rare. Fluid–fluid levels also have been described.29 Likewise, Liu et al.28 described occasional internal patchy enhancement of the lesion. Given the increased likelihood of sarcoma with contrast enhancement, these lesions may then not be classified as determinate and must be biopsied.28,30 If biopsy is considered, it is important to confirm that the lesion is not a vascular lesion, such as a pseudoaneurysm or arteriovenous malformation, and that the patient does not have an untreated coagulopathy. The diagnoses of pseudoaneurysm and arteriovenous malformation can often be made via a duplex ultrasound examination. Fig. 15.7 Hematoma. Sagittal T1-weighted (A) and sagittal fat-suppressed T2-weighted (B) images of the leg show a mass lesion in the anterior compartment (arrow in B). The T1-weighted image shows areas of hyperintensity (arrowheads), reflecting methemoglobin. The high signal intensity areas on the fat-suppressed T2-weighted image are related to soft-tissue edema and hemorrhage. Clinical correlation is especially helpful for the diagnosis of hematoma, and attention should be given to the presence or absence of coagulopathy, history of surgery, or other trauma. (From Papp DF, Khanna AJ, McCarthy EF, Carrino JA, Farber AJ, Frassica FJ. Magnetic resonance imaging of soft tissue tumors: determinate and indeterminate lesions. J Bone Joint Surg Am 2007;89(suppl 3):103-115. Reprinted by permission.) Heterotopic ossification is the formation of extraskeletal, mature, lamellar bone. This process most often occurs after direct trauma, as a complication of orthopaedic procedures and spinal cord injury, and in the burn patient. In post-traumatic cases, the process is termed myositis ossificans. Myositis ossificans occurs at sites of previous hematoma formations, although the process by which a hematoma involutes or evolves into myositis ossificans or a chronically expanding hematoma is not fully understood. Most patients present in the third decade of life, and the most common locations include the quadriceps and the brachialis muscles.31,32 The process is thought to arise after a direct impact to the affected muscle; the more severe the injury, the higher is the likelihood of hematoma formation.33 Radiographs show a well-circumscribed, calcified lesion in the pattern of mature, lamellar bone peripherally when the lesion has matured.15 In such cases, a conventional radiograph may be sufficient for diagnosis. However, when the diagnosis is in doubt, MRI can be helpful. T1-weighted images often show a lesion in the belly of the muscle, with a signal intensity the same as or slightly higher than that of the adjacent muscle. At times, this similarity may lead to the recognition of the lesion on T1-weighted images solely by noting the distortion of fascial planes.32 For lesions in patients who present early in their course, T2-weighted images show central high signal intensity with an external ring of low signal intensity. This configuration is pathognomonic and represents the zonal pattern of growth of myositis ossificans in which the external edges of the lesion ossify first. Surrounding edema may or may not be seen (Fig. 15.8). The diagnosis of myonecrosis should be considered when a patient presents with a rapidly growing, painful mass that involves at least one extremity. Patients who present with myonecrosis most frequently have involvement of the lower extremities, especially the quadriceps and calf muscles. Diabetes is the most frequently associated cause, but myonecrosis also has been associated with alcohol abuse and other, less common, entities. Diabetic myonecrosis has been reported in a previously healthy woman as a presenting symptom of her diabetes,34 although patients with diabetes more often have other existing sequelae, such as nephropathy or other forms of end-organ damage.35,36 Laboratory values are typically within normal limits, although elevated creatinine serum kinase levels have been reported.34,36 The ultimate pathophysiology of the process is not fully understood; it is best presented as a mixture of activated coagulation factors, impaired fibrin degradation, and endothelial damage from diabetic microangiopathy.37 These factors ultimately overcome the skeletal muscle’s abundant blood supply, leading to necrosis. Fig. 15.8 Myositis ossificans. (A) An axial T1-weighted image of the right leg shows a lesion (arrows) with the same intensity as the surrounding muscle, making the lesion difficult to see. The disruption of fascial planes is the only indication of abnormality. (B) An axial STIR image shows that the lesion (arrows) has a higher signal intensity than the surrounding muscles. Absence of a low signal intensity ring identifies it as an early lesion. Conventional radiographs with a zonal pattern of calcification aid with the diagnosis. The early recognition of myonecrosis, before tissue biopsy, can be important because patients with diabetes frequently have wound-healing problems. For this reason, nonsurgical management approaches are preferred.38 Conventional radiographs and CT imaging provide little data about the nature of this entity; however, MRI provides valuable information that facilitates diagnosis. T1-weighted imaging shows swelling and disruption of the involved muscles along the fascial planes. The muscle fiber pattern persists after the necrosis occurs, and there is no infiltration of the fascia. Although the resultant loss of striations is seen grossly on MRI, the overall structure remains unchanged. T2-weighted imaging shows a diffuse increase in signal intensity in the muscle, indicative of edema, and areas of necrosis. The muscles show heterogeneous signal intensity on T2-weighted images, which likely corresponds to fiber regeneration. The findings with myonecrosis contrast with those seen with neoplasms, which often disrupt the surrounding anatomy. Contrast-enhanced images may show a mixture of enhancing linear or serpentine fibers within low signal intensity regions. The dark (low signal intensity) areas represent necrotic tissue, and the enhancing areas represent viable or inflamed tissue (Fig. 15.9). The lesions frequently show peripheral enhancement on postgadolinium T1-weighted images.34,36,39 Neurofibromatosis type 1, formerly known as von Recklinghausen disease, is an autosomal dominant disorder characterized by neurofibromas and other systemic complications, such as the following: • Skeletal dysplasias • Café-au-lait spots • Lisch nodules • Vascular malformations • Learning disabilities • Optic gliomas In addition, these patients are at risk for other malignancies, such as nerve-sheath tumors and rhabdomyosarcoma, as well as for dedifferentiation of their neurofibromas into neurofibrosarcomas.40 With an incidence of approximately 1 in 3500, neurofibromatosis type 1 is more common than neurofibromatosis type 2 and is associated with the formation of true neurofibromas; the formation of schwannomas is more characteristic of neurofibromatosis type 2.40 Fig. 15.9 Myonecrosis. Axial T1-weighted (A) and T2-weighted (B) images of the right leg show a well-defined area (arrows) of low signal within the muscles of the anterior compartment of the leg in a patient with a history of diabetes mellitus. Note that the surrounding fascia and tissue planes are well preserved. On examination, the lesions can be superficial or deep and are not always painful. Despite their association with nerves, neurologic findings are uncommon. The MRI findings of neurofibromatosis are quite specific and therefore can help with diagnosis. The T1-weighted images show a lesion that is hyperintense relative to skeletal muscle. The appearance is fascicular or nodular. T2-weighted imaging classically shows a target sign, manifesting as an area of low signal centrally situated within a high signal intensity lesion. The myxoid nature of the neurofibroma accounts for the bright periphery, whereas the dark central region represents the compressed nerve fibers41,42 (Fig. 15.10). This specific finding simplifies the diagnosis and allows for ease of continued follow-up of the multiple lesions, which can help monitor for conversion to a neurofibrosarcoma. Superficial lesions do not always present with a target sign, can appear homogeneous, and usually extend to the skin.42 The presentation of a muscle tear masquerading as a soft-tissue mass occurs often enough that the orthopaedic surgeon should consider this possibility when addressing unknown soft-tissue lesions. Muscle tears commonly have a specific inciting incident; patients often remember hearing a snapping sound or feeling that their strength is “giving way,” followed by a period of muscle swelling, tenderness, ecchymosis, and/or edema. However, this scenario does not always occur. In fact, a study at the Walter Reed Army Medical Center showed that, in six of seven patients with rectus femoris muscle tears presenting as soft-tissue masses, there was no specific event; rather, the tear appeared insidiously, and only half of the lesions were painful.43
 Background
Background
 Soft-Tissue Tumors
Soft-Tissue Tumors
Determinate Lesions
Lipoma
Hemangioma
Ganglion Cyst
Synovial Cyst (Baker Cyst)
Hematoma
Myositis Ossificans
Myonecrosis (Diabetic and Idiopathic)
Neurofibroma
Muscle Tear
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


