Outline
Maternal Infections, 542
Fetal Cytomegalovirus Infection, 542
Fetal Parvovirus B19 Infection, 543
Fetal Rubella Syndrome, 544
Fetal Syphilis, 545
Fetal Toxoplasmosis Syndrome, 546
Fetal Varicella Zoster, 547
Teratogens, 548
Fetal Alcohol Syndrome/Fetal Alcohol Effects, 548
Fetal Valproic Acid Syndrome, 550
Fetal Warfarin Syndrome, 551
Maternal Diabetes/Caudal Regression Syndrome, 551
Central Nervous System, 553
Aicardi Syndrome, 553
Meckel Syndrome, 554
Lissencephaly, 556
Septo-optic Dysplasia, 557
Tuberous Sclerosis, 558
L1 Syndrome, 560
Cranium/Face, 561
Musculoskeletal System , 573
Other Lethal Skeletal Dysplasias, 580
Vertebral Anomalies, 583
Upper Extremity Anomalies, 585
Lower Extremity Anomalies, 589
Thorax, 590
Congenital High Airway Obstruction Syndrome, 590
Cornelia de Lange Syndrome, 592
Donnai-Barrow Syndrome, 592
Fryns Syndrome, 592
Pallister-Killian Syndrome, 593
Scimitar Syndrome, 594
Heart, 594
22q11.2 Deletion Syndrome, 595
Alagille Syndrome, 596
CHARGE Syndrome, 597
Heterotaxy Syndromes, 598
Holt-Oram Syndrome, 601
Noonan Syndrome, 601
Pentalogy of Cantrell, 603
Tuberous Sclerosis, 605
Gastrointestinal System/Abdominal Wall, 605
Genitourinary Tract, 611
Abnormal Fetal Movement, 613
Antley-Bixler Syndrome, 613
Caudal Regression Syndrome, 613
Congenital Ichthyosis, 613
Congenital Myasthenic Syndromes, 614
Congenital Myotonic Dystrophy, Type 1, 614
Fetal Akinesia, 615
Freeman-Sheldon Syndrome, 617
Prader-Willi Syndrome, 617
Fetal Overgrowth, 618
Bannayan-Riley-Ruvalcaba Syndrome, 618
Beckwith-Wiedemann Syndrome, 607
Klippel-Trenaunay Syndrome, 619
Megalencephaly-Polymicrogyria-Polydactyly-Hydrocephalus Syndrome, 619
Perlman Syndrome, 620
Proteus Syndrome, 620
Simpson-Golabi-Behmel Syndrome, 621
Sotos Syndrome, 622
Weaver Syndrome, 622
Fetal Growth Restriction, 623
Microdeletion Syndromes, 624
Deletion 4p (Wolf-Hirschhorn Syndrome), 624
Deletion 5p (Cri du Chat Syndrome), 625
Deletion 11q (Jacobsen Syndrome), 625
Deletion 22q11.2 (DiGeorge Syndrome), 626
Metabolic Syndromes, 626
Other Malformations, 628
Summary of Key Points
- •
Congenital anomalies can occur in isolation or be indicative of a more extensive underlying process.
- •
The presence of a congenital anomaly should prompt a thorough evaluation for other anomalies and consideration of additional advanced fetal imaging.
- •
When an underlying global process is suspected, genetic testing is indicated and a geneticist or genetic counselor should be involved.
- •
With advances in molecular diagnostics, fetal syndromes are more readily diagnosed by use of both chromosomal microarray and next-generation sequencing technologies.
- •
When fetal syndromes are diagnosed prenatally, a multidisciplinary team should assist in counseling and care coordination for expectant parents.
- •
Confirmation of an underlying genetic alteration leading to the observed fetal anomalies can both assist in diagnosis as well as provide information crucial for future reproductive planning.
Prenatally, a congenital anomaly is suspected when images of a fetal organ differ from the expected development for a given gestational age. A congenital anomaly can result from a malformation (an intrinsically abnormal developmental process), deformation (normal development with extrinsic forces), disruption (breakdown of previously normal tissue), or dysplasia (an abnormal organization of cells within tissues which may disrupt more than one organ) ( Fig. 16-1 ). Whenever one congenital anomaly is present, a thorough search for other anomalies and unusual findings (i.e., accelerated or restricted fetal growth, abnormal amniotic fluid levels, altered fetal movements) should be undertaken. Additional imaging modalities may be helpful in completing the full fetal assessment including three-dimensional (3D) ultrasound studies, Doppler interrogations, and fetal magnetic resonance imaging (MRI).
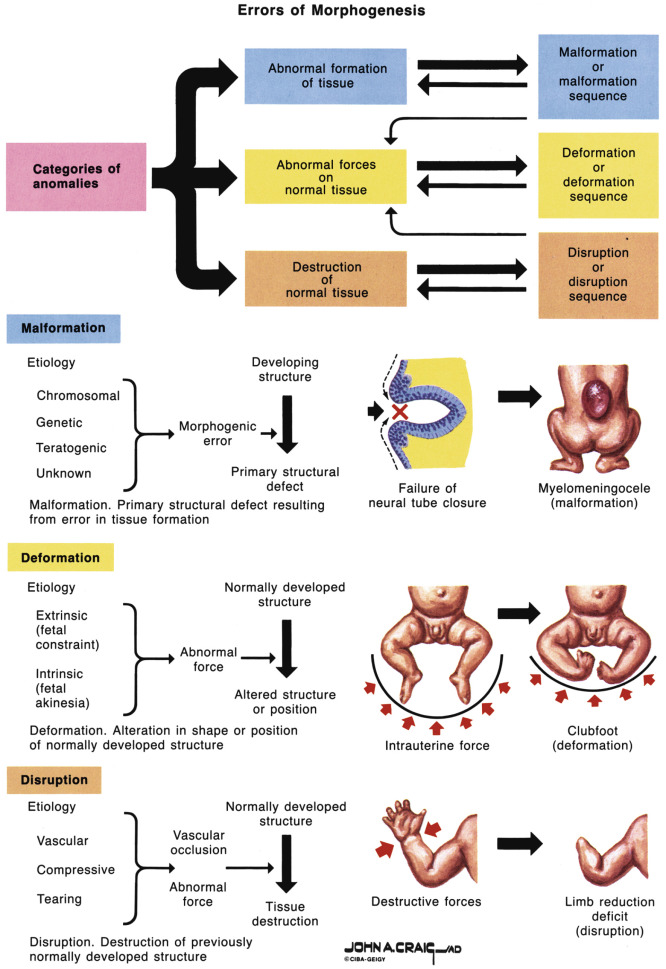
The presence of congenital anomalies should prompt consideration of underlying global disorders, especially when several imaging findings are present. Multiple findings may be the result of a sequence, syndrome, or association. In a sequence, a group of anomalies is caused by an initial malformation, deformation, or disruption (i.e., amniotic band sequence). In a syndrome, a group of anomalies is the result of an underlying genetic cause and is pathogenetically related (i.e., Down syndrome). In an association, a given set of anomalies occurs together more frequently than can be explained by chance and the particular anomalies are not known to be the result of a sequence or syndrome (i.e., VACTERL association [ v ertebral anomalies, a nal atresia, c ardiac anomalies, t racheoesophageal fistula or e sophageal atresia, r enal/urinary anomalies, and l imb defect]).
As molecular diagnostic technologies have become more refined and accessible, fetal syndromes are more readily diagnosed in both prenatal and postnatal settings. Prenatally, when a congenital anomaly is seen, chromosomal microarray is now recommended as the first-line test. In addition to aneuploidy, chromosomal microarray allows detection of microdeletion and microduplication syndromes and leads to the identification of approximately 5% more cytogenetic abnormalities than can be seen on conventional karyotype alone ( Fig. 16-2 ). However, many syndromes are single gene disorders and cytogenetic testing by both karyotype and chromosomal microarray will be normal. For many of these disorders, individual gene testing and gene panel testing is now available. Cost and time to obtain fetal DNA results prenatally still remain a significant hurdle for many syndromes suspected on prenatal imaging. The prenatal diagnostician should work closely with neonatal care providers to assure completion of a genetic workup.

In the following pages, we review fetal syndromes that are potentially detectable based on prenatal imaging findings and group them by prominent features (either the body system affected or some other hallmark such as altered fetal growth or movement). The review is not exhaustive but serves to highlight fetal syndromes commonly encountered by sonologists and sonographers and presents relevant imaging findings and genetics. Fetal syndromes often come to initial clinical attention based on screening prenatal ultrasound imaging; further imaging with 3D sonography and fetal MRI (especially for evaluation of the central nervous system [CNS]) can aid in delineation of additional features. Other specialties, including genetics, maternal-fetal medicine, and pediatric subspecialties (i.e., cardiology, surgery), can then assist with specialized testing, obstetric care, and postpartum planning for patients continuing with their pregnancies. These providers can give a clear overview of the anticipated process to the parents and convey the time and complexities of reaching a specific diagnosis for the fetus or child.
Given the rapid evolution in genetic testing, involvement of the genetics team is particularly important to assure that the most appropriate diagnostic testing is performed either in the prenatal or postnatal period. For many single gene disorders, multiple causative mutations within one gene have been identified. At times, the mutation may be unique or “private” to the family themselves. Completion of genetic studies provides not only confirmation of the diagnosis but an opportunity for the family to utilize the information in future reproductive planning.
Maternal Infections
Fetal Cytomegalovirus Infection
Definition
Fetal cytomegalovirus (CMV) infection is a congenital disorder characterized by hydrops, ascites, and ventriculomegaly caused by transplacental transmission of CMV to the fetus. The double-stranded DNA herpes group virus causes a mild infection or a mononucleosis-type illness in young healthy adults, chronic disease in older adults, and mild to severe congenital infection. Congenital infection is mostly caused by maternal primary infection.
Synonym
Fetal cytomegalovirus infection is also known as congenital cytomegalovirus infection.
Etiology
Cytomegalovirus (a double-stranded DNA herpes group virus) is the cause of the infection.
Incidence
Congenital CMV infection occurs in approximately 1% of all deliveries and is the most common congenital infection. Intrauterine transmission of CMV takes place in approximately 40% of infections, and approximately 10% of liveborn infants have symptomatic disease at the time of birth and later.
Diagnosis
CMV infection, as well as other congenital infections, should be suspected whenever nonimmune hydrops is found. Other suggestive findings include intracranial calcifications and intracranial hemorrhage, microcephaly, brain atrophy, abnormal periventricular echogenicities, intraparenchymal foci, ventriculomegaly, intraventricular adhesions, periventricular pseudocysts, sulcation and gyral abnormal patterns, hypoplastic corpus callosum, cerebellar and cisterna magna abnormalities, signs of striatal artery vasculopathy, splenomegaly, chorioretinitis, occlusion of the foramen ovale, signs of right-sided heart overload from the premature closure, ascites, hyperechoic bowel, fetal growth restriction, and oligohydramnios. Most features can be found by sonography beginning at approximately 20 weeks’ gestation. Whenever maternal infection is confirmed, polymerase chain reaction (PCR) testing of amniotic fluid should be performed. PCR testing of amniocentesis samples should be performed after a 6-week interval following the diagnosis of maternal infection and is most sensitive after 21 weeks’ gestation. The diagnosis can also be made by histologic study of typical inclusion bodies in biopsy or autopsy specimens. Focal sonographic periventricular increased echogenicity associated with mild ventriculomegaly, without any abnormalities of the cerebral and cerebellar organogenesis or cephalic biometry alteration in the third trimester of pregnancy, should be considered a marker of encephalitis following CMV infection. Fetal MRI is a useful adjunct in the evaluation of intrauterine infection with CMV.
Pathogenesis
The exact mode of transplacental passage is uncertain. The virus replicates in fetal tissues, producing inflammation, tissue necrosis, and organ dysfunction. CMV hepatitis in the neonate can present with an intense inflammatory response involving the portal triads. In these cases, lobular disarray, degeneration of hepatocytes, and cholestasis are also seen. The cause of ascites in congenital CMV infection is not certain. Contributing factors may include low serum protein levels due to hepatic dysfunction and portal obstruction resulting from periportal inflammation.
Associated Anomalies
Isolated ascites is an uncommon finding in fetuses with CMV infection but may occur. Cardiovascular, gastrointestinal (GI), musculoskeletal, and ocular lesions may be found in association with the classic features. Petechiae, sensorineural hearing loss, and poor intellectual development may also occur after birth.
Differential Diagnosis
Because ascites is often the first manifestation of hydrops, the differential diagnosis for fetal ascites is essentially the same as with generalized hydrops, which includes many congenital infections. Conditions that present with intracranial calcifications (such as tuberous sclerosis), hyperechoic bowel (cystic fibrosis and Down syndrome), and hepatomegaly (primary liver disease or extramedullary hematopoiesis) should be considered.
Prognosis
In general, neonates with symptomatic CMV infection do poorly, with a neonatal mortality rate of 5%, and 50% to 60% of survivors suffer severe long-term neurologic morbidity. CMV hepatitis is reversible in survivors, but intellectual disability, motor deficits, and hearing loss are expected long-term sequelae. Late sequelae, such as sensorineural hearing loss and neurodevelopmental disorders, occur in 10% to 15% of infants lacking symptoms at birth. Pediatric neurologic morbidity is related to the degree of antenatal ventriculomegaly and, when greater than 15 mm, is associated with an increase in abnormal neurologic development. A normal ultrasound examination does not exclude the possibility of symptoms in the newborn or long-term complications.
Recurrence Risk
Given that viral infection confers immunity in most patients, there is only a small theoretical risk of reinfection in another pregnancy.
Management
Termination of pregnancy can be offered before viability. If continuation of the pregnancy is chosen, follow-up with ultrasound examinations every 2 to 4 weeks is recommended to monitor for growth restriction, hydrops, and other fetal manifestations. Antiviral medications have not been shown to decrease the rate of perinatal transmission. Trials are under way for treatment of pregnant women with CMV infection early in pregnancy with hyperimmunoglobulin therapy.

Fetal Parvovirus B19 Infection
Definition
Infection with parvovirus B19 causes erythema infectiosum and is a common childhood illness characterized by a “slapped cheek” facial appearance and a lacy erythematous rash on the trunk and extremities. Patients may have systemic symptoms prior to the appearance of the rash, including arthropathy.
Synonym
Fetal parvovirus B19 infection is also known as fifth disease.
Etiology
Acute maternal parvovirus B19 viremia leads to transplacental passage of the B19 virus and subsequent fetal infection.
Incidence
Acute parvovirus B19 infection occurs in 3% to 4% of pregnant women, with the highest infection rates in school teachers and day care workers. Approximately 35% to 53% of pregnant women have immunity from a prior infection.
Pathogenesis
The interval between maternal infection and fetal hydrops is about 3 weeks, and 93% of cases of fetal hydrops occur within 8 weeks of maternal diagnosis. Acute infection can lead to fetal loss or hydrops fetalis. Hydrops occurs because parvovirus B19 preferentially infects rapidly dividing cells and is cytotoxic to fetal red blood cell precursors, leading in some cases to severe fetal anemia and subsequent hydrops. Parvovirus B19 can also infect myocardial cells and cause myocarditis; this infection may also initiate apoptosis in cells. The risk of hydrops is greater if infection occurs in the first half of pregnancy. Hydrops can either lead to rapid fetal death or resolve spontaneously (although the rate of spontaneous resolution is uncertain).
Associated Anomalies
In case reports, other congenital abnormalities have been noted, including ocular abnormalities, hydrocephalus, cleft lip/palate, joint webbing, musculoskeletal anomalies, hepatocellular damage, myocarditis, congenital cardiomyopathy, and myositis.
Diagnosis
Acute maternal parvovirus infection can be diagnosed by positive IgM serologic findings; IgM can be detected 10 days after infection and may persist for 3 months or longer. IgM can be falsely negative, however, and failure to detect IgM antibodies therefore does not rule out B19 infection. Parvovirus B19 IgG develops several days after IgM and indicates past infection. Maternal B19 viremia can also be confirmed by PCR with improved sensitivity. Fetal parvovirus infections can be confirmed by PCR for B19 DNA in amniotic fluid.
Differential Diagnosis
Differential diagnosis includes other conditions leading to nonimmune hydrops in the fetus.
Prognosis
Risk of fetal death is largely limited to parvovirus B19 infections diagnosed in the first half of pregnancy (13% in first trimester, 9% from weeks 13 to 20, and <1% after 20 weeks). Intrauterine transfusion of hydropic fetuses improves prognosis. In addition to fetal anemia, severe fetal thrombocytopenia can occur and lead to exsanguination at the time of intrauterine red blood cell transfusion. Children who survive fetal hydrops generally do well; an increased risk of developmental delay has been reported, but this has not been demonstrated in all studies.
Recurrence Risk
There is virtually no recurrence risk because pathogenesis occurs with primary maternal parvovirus B19 infection.
Management
Pregnant women exposed to parvovirus should have serologic testing for B19 IgM and IgG; positive IgG indicates maternal immunity, and the fetus should be protected. A positive IgM antibody indicates acute infection. Fetal intervention prior to 18 to 20 weeks is often not feasible, and transfusions this early in gestation are associated with an increased risk for fetal loss. Women diagnosed after 20 weeks should receive weekly ultrasound examinations to assess for fetal anemia and hydrops for at least 8 weeks after infection. If severe anemia is detected, intrauterine fetal blood transfusion is indicated.
Fetal Rubella Syndrome
Definition
Fetal rubella syndrome is a congenital disorder resulting from primary maternal infection with the rubella virus. It is characterized by deafness, mental retardation, congenital cataract, heart defects, and other structural anomalies found with variable severity and frequency. The rate of congenital infection with acute rubella during pregnancy is higher than 90% in the first 12 weeks of pregnancy, approximately 60% in weeks 13 to 17, 25% in weeks 18 to 24, and increases again during the last month of pregnancy.
Synonyms
Synonyms for fetal rubella syndrome are fetal rubella effects, congenital rubella syndrome, and German measles.
Etiology
Fetal rubella is caused by an RNA togavirus, which is the only member of the genus Rubivirus . The fetus is infected by transplacental transmission followed by hematogenous spread.
Recurrence Risk
There is no recurrence risk of fetal rubella syndrome.
Incidence
Cases of fetal rubella syndrome are rare in the United States since 2004 following comprehensive vaccination; however, reinfection after vaccination is possible. There were 13 cases reported in the United States between 2001 and 2004. It remains a clinical issue in other parts of the world.
Diagnosis
The most frequent sonographic findings are cardiac malformations (in particular, septal defects), eye defects (cataracts, microphthalmia, and retinopathy), microcephaly, hepatomegaly, splenomegaly, and growth restriction. The risk of congenital defects is limited to maternal infection during the first 16 weeks of pregnancy. Sensorineural hearing loss and developmental delay are also associated with congenital rubella sydrome. The confirmation of fetal infection can be made by isolating rubella viral RNA from amniotic fluid samples and testing using PCR.
Associated Anomalies
Occasionally, the following anomalies can be associated with the classic findings of fetal rubella syndrome: renal disorders, hypospadias, cryptorchidism, meningocele, glaucoma, patent ductus arteriosus, and peripheral pulmonary stenosis.
Differential Diagnosis
Other conditions associated with congenital hepatomegaly and cataracts should be considered. This includes other congenital infections (TORCH [ t oxoplasmosis, o ther infections, r ubella, c ytomegalovirus, h erpes simplex]), fetal anemia, fetal liver tumors, chondrodysplasia punctata, Neu-Laxova syndrome (NLS), Smith-Lemli-Opitz (SLO) syndrome, and Walker-Warburg syndrome.
Prognosis
Spontaneous abortion and intrauterine death may occur. The postnatal impact of intrauterine infection varies from absence of any defect to all the anomalies mentioned earlier with variable severity. The virus may remain for years in body tissues, causing complications of chronic infection (such as diabetes mellitus from chronic viropathy of the pancreas).
Management
Termination of pregnancy should be discussed when fetal infection is detected during the first trimester, owing to the severity of the condition in this group. After viability, monthly ultrasound examinations for growth and follow-up of the anomalies are recommended.
Prevention
Women found to be rubella nonimmune during pregnancy should be offered vaccination postpartum prior to discharge from the hospital. Breastfeeding is not a contraindication to vaccination.
Fetal Syphilis
Definition
Fetal syphilis is caused by fetal infection with the spirochete Treponema pallidum, which readily crosses the placenta leading to fetal infection.
Synonym
A synonym for fetal syphilis is congenital syphilis.
Etiology
Maternal infection with T. pallidum leads to transplacental transmission of the virus and resultant fetal infection.
Incidence
Incidence in 2008 was 10.1 cases per 100,000 liveborn infants with many cases found in women without prenatal care. The risk of transmission is higher for primary and secondary syphilis (50%) than for early latent (40%) and late (10%) syphilis. Transmission can occur at any gestational age. The frequency of vertical transmission increases as gestation advances but severity decreases with infection later in pregnancy.
Diagnosis
In women with positive syphilis testing, clinical findings concerning for congenital syphilis include growth restriction, as well as liver and placental findings. Signs of liver dysfunction include hepatomegaly and ascites, which can lead to nonimmune hydrops. The placenta is typically large and edematous. Silver-staining of the placenta following delivery is positive for spirochetes. Fetal serologic tests are positive for antitreponemal IgM.
Associated Anomalies
Clinical findings in neonates may include the following:
- •
Early congenital syphilis: hepatomegaly, syphilitic rhinitis (“snuffles”), maculopapular rash, generalized lymphadenopathy, and skeletal abnormalities.
- •
Late congenital syphilis (generally a result of gumma formation in various tissues): facial features (frontal bossing, saddle nose, short maxilla), eye findings (interstitial keratitis, glaucoma, corneal scarring, optic atrophy), sensorineural hearing loss, Hutchinson teeth, mulberry molars, perforation of hard palate, rhagades, gummas, anterior bowing of shins (“saber shins”), and paroxysmal cold hemoglobinuria.
Differential Diagnosis
Findings in neonates can resemble other congenital infections.
Prognosis
The severity of fetal effects depends on the duration of infection as well as the stage of development at the time of infection.
Recurrence Risk
Recurrence is likely only if maternal syphilis infection remains untreated.
Management
All women presenting for prenatal care should be evaluated for syphilis infection. If positive, penicillin treatment should be given; regimens vary according to the chronicity of the disease. If the duration of the disease is unknown, three doses of benzathine penicillin given at weekly intervals is recommended. If maternal syphilis is treated at least 30 days prior to delivery, only 1% to 2% of infants will be infected. This rate is greater than 70% if maternal syphilis remains untreated. If maternal syphilis is diagnosed after 20 weeks’ gestation, an ultrasound examination should be obtained to assess for congenital syphilis.

Fetal Toxoplasmosis Syndrome
Definition
Toxoplasmosis is caused by infection with the protozoan parasite Toxoplasma gondii . Toxoplasmosis is normally asymptomatic in immunocompetent individuals. Acute infections in pregnant women can be transmitted to the fetus and cause severe illness (mental retardation, blindness, and epilepsy). The risk of maternal-fetal transmission increases with gestational age at the time of exposure, whereas the incidence of severe disease decreases.
Synonym
Fetal toxoplasmosis syndrome is also known as congenital toxoplasmosis.
Incidence
An estimated 400 to 4000 cases of congenital toxoplasmosis occur annually in the United States. Of the 750 deaths attributed to toxoplasmosis each year, 375 (50%) are believed to be caused by eating contaminated meat, making toxoplasmosis the third leading cause of food-borne deaths in the United States. The incidence of toxoplasmosis acquisition during pregnancy ranges from 1 to 4 per 10,000. Half of fetuses escape the infection, one third have subclinical infection, and about 10% have severe infection.
Etiology
Fetal toxoplasmosis is caused by transplacental transmission of parasites following maternal primary infection. Transmission from mothers with chronic infection via reactivation is rare but can be caused by immunologic dysfunction. The rate of fetal transmission increases greatly as the gestational age at time of seroconversion increases, with transmission rates of 25%, 54%, and 65%, in the first, second, and third trimesters, respectively.
Pathogenesis
The fetus is infected hematogenously through the placenta during parasitemia in the mother. Ocular toxoplasmosis causes irreversible damage to the retina in utero. The fetus and infant mount inflammatory responses that may contribute to ocular damage.
Diagnosis
The classic triad suggestive of congenital toxoplasmosis includes chorioretinitis, intracranial calcifications, and hydrocephalus. Less common findings include ascites, pericardial and pleural effusions, and intrahepatic densities. However, most infants infected in utero are born with no obvious signs of toxoplasmosis on routine examination, but many develop learning and visual disabilities later in life. If left untreated, congenital toxoplasmosis can be associated with severe and even fatal disease. Other findings include microcephaly, encephalomyelitis, seizures, intellectual disability, ascites, and hepatosplenomegaly. The diagnosis can be made by using PCR to detect of Toxoplasma gondii in amniotic fluid, with increased sensitivity later in pregnancy.
Differential Diagnosis
Other TORCH infections should be considered.
Prognosis
Approximately 75% of congenitally infected newborns are asymptomatic.
Recurrence Risk
There is typically no recurrence risk.
Management
Depending on gestational age and whether the fetus is known to be infected, pregnant women have been treated with the antibiotic spiramycin or with sulfadiazine alone or the combination of pyrimethamine and sulfadiazine. Treatment of acute infection during pregnancy has been associated with an approximately 50% reduction in fetal infection.
Prevention
Toxoplasmosis infection can be prevented in large part by cooking meat to a safe temperature and peeling or thoroughly washing fruits and vegetables before eating. Pregnant women should avoid changing cat litter or, if no one else is available to change the cat litter, use gloves, then wash their hands thoroughly.


Fetal Varicella Zoster
Definition
Fetal varicella zoster is characterized by abnormalities of multiple organs caused by fetal infection with varicella following maternal chickenpox infection.
Synonyms
Fetal varicella zoster is also known as congenital varicella syndrome, varicella embryopathy, and chickenpox.
Incidence
The incidence of maternal varicella infection was 1 to 5 in 10,000 pregnancies in the United States in the 1980s but has decreased since vaccination began in 1995. The risk of fetal involvement among all pregnant women infected with varicella varies from 1% to 20%. First trimester varicella infections have been associated with an increased risk of spontaneous abortion. Second trimester varicella infections have been associated with 2% risk of a congenital syndrome characterized by limb hypoplasia, cutaneous scars, cataracts, microcephaly, and cortical atrophy.
Etiology
Herpesvirus is the etiologic agent.
Recurrence Risk
Recurrence risk is less than 1%.
Diagnosis
Maternal infection at any time in pregnancy exposes the fetus to a high risk of transplacental contamination and warrants follow-up. The risk of fetal anomalies, however, is the greatest from 8 to 20 weeks’ gestation. Sonographic signs of fetal disease include fetal demise, growth restriction, musculoskeletal abnormalities (such as clubfeet and abnormal position of the hands, caused by both necrosis and denervation of the affected tissue), limitation of limb extension due to cicatrices formation, cutaneous scars, limb hypoplasia, chorioretinitis, congenital cataracts, microphthalmia, hydrops, polyhydramnios, hyperechogenic hepatic foci, cerebral anomalies (such as ventriculomegaly or atrophy and microcephaly), disseminated foci of necrosis and microcalcifications, encephalitis, and echogenic bowel in the second trimester. The placenta can show a multifocal chronic villitis with multinucleated giant cells. Fetal infection can be demonstrated by detection of varicella zoster virus DNA by PCR in fetal blood and amniotic fluid.
Pathogenesis
Maternal viremia leads to placental infection with subsequent fetal transmission. Direct damage occurs to fetal tissues by the neurotropic varicella virus.
Associated Anomalies
Congenital anomalies in multiple organs with variable severity can be seen in varicella embryopathy. Among survivors, intellectual disability, seizures, and limitation of movements may develop after birth. The virus may cause serious infections, particularly pneumonia, in adult women.
Differential Diagnosis
Other viral infections, vascular accidents, and amniotic band syndrome should be considered.
Prognosis
The severity of fetal involvement varies from dermatologic lesions to lethal disseminated disease. Limited scarring tends to have an excellent prognosis. Fetal brain disruptions, or severe maternal varicella with development of lethal maternal pneumonia and encephalitis, are indicators of an extremely high risk for fetal demise. The rate of fetal demise varies from 39% to 61%. Maternal infection in the first and early second trimester has a higher association with fetal anomalies, whereas third trimester infections have a higher risk for varicella zoster development at the neonatal period. A life-threatening illness may occur in the newborn when delivery occurs within 5 days of the onset of maternal illness.
Management
Termination of pregnancy can be offered before viability. If continuation of the pregnancy is chosen, ultrasound follow-up is recommended to assess for fetal anomalies, limb contractures, and other signs of fetal compromise. If maternal seroconversion is suspected for varicella zoster, prenatal sonography and MRI may document the extent of tissue damage and assist in counseling. After a therapeutic abortion, fetal infection can be confirmed by detection of varicella zoster viral DNA in fetal tissues and the placenta, as well as by histopathologic findings such as miliary calcified necroses in fetal organs.
Prevention
Serologic testing and vaccination should be offered to women of childbearing age, and women should be questioned prior to conception about varicella immunity. Susceptible pregnant patients should be counseled to avoid contact with individuals who have chickenpox. If exposure occurs, varicella zoster immunoglobulin should be administered within 96 hours in an attempt to prevent maternal infection. Susceptible neonates should also receive varicella zoster immunoglobulin. Acyclovir is active against the varicella zoster virus, and treatment is indicated in seriously ill adults and neonates.

Teratogens
Fetal Alcohol Syndrome/Fetal Alcohol Effects
Alcohol use during pregnancy results in a spectrum of adverse outcomes known as fetal alcohol spectrum disorders. Fetal alcohol syndrome (FAS) is one of these disorders. FAS is characterized by specific facial abnormalities and significant impairments in neurodevelopment and physical growth. Children exposed to alcohol (approximately 45 to 50 g of ethanol per day or equivalent) in utero suffer from growth and mental retardation, physical abnormalities, and immune dysfunction. There is no “threshold,” so some fetuses exhibit signs of fetal alcohol effects at lower exposure. Recommendations for clinicians regarding assessment of thresholds published by the National Institute on Alcohol Abuse and Alcoholism recommend that any woman who reports drinking more than seven drinks per week or more than three drinks on any given day be further assessed for risk of developing alcohol-related problems.
Synonyms
FAS is one of the fetal alcohol spectrum disorders.
Incidence
The incidence of FAS ranges from 2 to 30 per 10,000 live births. FAS is the most common cause of intellectual disability in the United States and is thought to occur in 0.5 to 2.0 in 1000 births. The true extent of teratogenic injury from alcohol exposure exceeds the clinically recognized prevalence of FAS, because behavioral and physical teratogenesis may be present in the absence of full expression of the syndrome.
Etiology/Pathogenesis
FAS is caused by direct toxicity of alcohol and its metabolites that cross the placenta and are not detoxified by the fetal liver. Alcohol is a teratogen and inflicts irreversible damage on the CNS. The rate of alcohol elimination from the fetal compartment is only 3% to 4% that of the maternal rate. Alcohol has fetal effects in all trimesters of pregnancy.
Diagnosis
The findings include microcephaly, long round philtrum, small micrognathia, cleft palate, suppression of the cupid arch, microphthalmia, microcephaly, dysgenesis of the corpus callosum, malformed ears, atrial septal defect (ASD), ventricular septal defect (VSD), and growth restriction predominantly involving the limbs and occurring early without oligohydramnios. This lack of specificity suggests that the FAS facies may be a common teratogenic expression of exposure to a variety of substances occurring during a defined period of fetal development.
Differential Diagnosis
Other conditions that involve growth restriction and microcephaly, such as congenital infections and chromosomal anomalies, should be considered.
Prognosis
Intellectual disability and delayed growth persist postnatally. Most children with FAS have mild to moderate intellectual disability, but this can vary widely. The severity of intellectual disability appears related to the severity of growth deficits and dysmorphogenesis, such that the more phenotypically affected individuals have lower IQ scores. Hyperactivity is frequently observed. As adults, psychiatric disorders are highly prevalent in individuals with FAS, affecting more than 70% in one series, in which 60% had alcohol or drug dependence, 44% depression, and 40% psychosis.
Recurrence Risk
Recurrence risk is high (up to 70%) in subsequent pregnancies if mothers continue to drink alcohol.
Management
The management of these pregnancies should be aimed at reducing alcohol consumption; few programs have had significant efficacy.



Fetal Valproic Acid Syndrome
Definition
Fetal valproic acid exposure syndrome results from maternal valproate (an anticonvulsant) use during pregnancy and is characterized by CNS dysfunction, spina bifida, developmental delay, fetal growth restriction, and cardiac anomalies.
Synonym
Depakote exposure is a synonym for fetal valproic acid syndrome.
Incidence
Fetal valproic acid syndrome is rare, and the incidence is unknown. Any epileptic pregnant mother has two to three times increased risk for congenital anomalies compared with the general population. If the exposure to valproic acid takes place between the 17th and 30th days after fertilization, the incidence of neural tube defects approaches 1% to 2%. In general, the teratogenic risks are higher with increasing doses of valproic acid, with a significantly higher rate of malformations in doses up to 1000 mg/day.
Etiology
Exposure to valproic acid is the cause of this syndrome.
Pathogenesis
The pathogenesis of fetal valproic acid syndrome is unknown.
Diagnosis
The findings include cardiovascular abnormalities, hypotonia, spina bifida, hypospadias, and limb reduction. The facial appearance can be characterized by an oral cleft, small broad nose, small ears, flat philtrum, a long upper lip with shallow philtrum, and micrognathia/retrognathia. Fetal growth restriction, microcephaly, generalized hypertrichosis sparing palms and soles, coarse face, gum hypertrophy, clubfeet and clubhands, musculoskeletal abnormalities, genital abnormalities, and urogenital defects may also be present. Heart defects have also been reported. In 26% of patients with fetal valproic acid exposure syndrome, cardiovascular abnormalities, most frequently VSDs, aortic or pulmonary stenosis, and persistent ductus arteriosus also occur. Pulmonary hypoplasia is also reported. Epilepsy and intellectual disability may develop after birth.
Associated Anomalies
Associated anomalies include omphalocele, inguinal hernia, duodenal atresia, and scoliosis. Hyperbilirubinemia, hepatotoxicity, transient hyperglycinemia, afibrinogenemia, and fetal or neonatal distress may be found.
Differential Diagnosis
Neural tube defects of alternative origin should be considered. However, the clinical history in the presence of spina bifida and cardiac anomaly should suggest the diagnosis.
Prognosis
Fetuses with major anomalies have a poor prognosis. Metabolic disturbances may also complicate the neonatal period. Affected children can die in infancy, and the surviving patients can have intellectual disability.
Recurrence Risk
If the mother is exposed to valproic acid in a second pregnancy, the teratogenic effect is likely to be similar.

Fetal Warfarin Syndrome
Definition
Fetal warfarin syndrome is characterized by specific bone and cartilage abnormalities in the fetus, including nasal hypoplasia, limb hypoplasia, and stippled epiphyses. It is caused by fetal exposure to warfarin between weeks 6 and 12 of gestation.
Synonyms
Fetal warfarin syndrome has also been described as warfarin embryopathy and coumadin exposure.
Incidence
A wide range has been reported, but the best estimate is that less than 10% of exposed fetuses will have embryopathy. The teratogenic effect is dose-dependent, with greater safety if the dose is less than 5 mg/day.
Etiology
Warfarin freely crosses the placenta and is a known teratogen, with the highest risk between weeks 6 and 12 of gestation.
Pathogenesis
The pathogenesis is unknown. Some hypotheses suggest that the drug interferes with post-translational modification of calcium-binding proteins essential for the development of bony structures.
Diagnosis
Common sonographic findings include developmental abnormalities of bone and cartilage, specifically nasal and limb hypoplasia and stippled epiphyses.
Associated Anomalies
CNS abnormalities have been more rarely reported and include microcephaly, optic atrophy, intellectual disability, hypotonia, and spasticity.
Differential Diagnosis
Differential diagnosis includes other disorders affecting the bone and cartilage, specifically chondrodysplasia punctata.
Prognosis
Because warfarin freely crosses the placenta, it can cause fetal bleeding at any gestational age. The most serious fetal complications are related to cerebral hemorrhage. Data on long-term survivors are lacking.
Recurrence Risk
The recurrence risk is unknown. Fetal warfarin syndrome is related to warfarin exposure in subsequent pregnancy.
Management
When detected prior to viability, termination of the pregnancy can be offered. If the pregnancy continues, no alteration in management is needed. If warfarin is continued throughout the pregnancy, it should be discontinued at 34 to 36 weeks’ gestation and an alternative agent should be used for anticoagulation to minimize the risk of fetal bleeding at the time of delivery.
Maternal Diabetes/Caudal Regression Syndrome
Definition
Caudal regression syndrome is a rare congenital defect, characterized by the absence of the sacrum, variable defects of portions of lumbar spine, and anomalies in other systems.
Synonyms
Caudal dysplasia sequence and sacral agenesis are other terms that have been used to describe caudal regression syndrome.
Incidence
The incidence of caudal regression syndrome is 0.25 to 1 per 10,000 normal pregnancies. This risk is 200 to 250 times higher in diabetic pregnancies.
Etiology
The cause is unknown, but caudal regression syndrome is associated with maternal diabetes in 16% of the affected fetuses.
Recurrence Risk
This anomaly is not thought to be hereditary, and the recurrence risk is very small, although it is higher in diabetics.
Diagnosis
The sonographic findings are variable, and depend on the extent and severity of the defect. It ranges from complete absence of the sacrum associated with abnormalities of the lumbar spine and lower extremities (such as clubfeet and contractions of the knees and hips) to abnormalities of the sacrum without associated defects. The most typical findings are the absence of a few vertebrae, the shield-like appearance of the fused or approximated iliac wings, and decreased interspace between the femoral heads. Some sonographic planes of section will intersect the fetus at such an angle that no spine is visible, a very striking and probably pathognomonic finding. Decreased movement of the legs is frequently observed. First trimester diagnosis may be difficult because of incomplete ossification of the sacrum at that time. A short crown-rump length and abnormal appearance of the yolk sac have been proposed as early sonographic signs of caudal regression syndrome.
Genetics
The genetics of caudal regression syndrome is unknown.
Pathogenesis
This syndrome is thought to be due to disrupted maturation of the caudal portion of the spinal cord complex before 4 weeks’ gestation, leading to motricity deficits and neurologic impairment, varying from incontinence of urine and feces to complete neurologic loss.
Associated Anomalies
Anomalies of the central nervous, musculoskeletal, genitourinary, cardiac, respiratory, and GI systems may be found in association.
Differential Diagnosis
Sirenomelia is the main alternative diagnosis and was thought to be the most severe form of caudal regression syndrome previously; it is now considered a separate entity. Fusion of the lower extremities is generally seen in sirenomelia.
Prognosis
Prognosis depends on the severity of the spinal defect and associated anomalies, but the vast majority of survivors require urologic and orthopedic intervention. Severe forms are commonly associated with cardiac, renal, and respiratory problems, which are responsible for early neonatal death.



Central Nervous System
Characteristic CNS abnormalities are seen in a majority of genetic syndromes, and, thus, an exhaustive list of associated syndromes is beyond the scope of this chapter. CNS abnormalities can be seen in all regions of the brain and include Chiari malformation, holoprosencephaly, lissencephaly, polymicrogyria, cerebellar hypoplasia, agenesis of the corpus callosum, hydrocephalus, neural tube defects, and CNS tumors. Many of these abnormalities and associated conditions are discussed in more detail in Chapter 9 , Ultrasound Evaluation of the Fetal Central Nervous System. In this chapter, several syndromes with prominent CNS findings will be reviewed in greater detail, including Aicardi syndrome, Meckel syndrome (MKS), lissencephaly-associated syndromes (Miller-Dieker, Walker-Warburg, Baraitser-Winter, Norman-Roberts, microlissencephaly, and Neu-Laxova syndromes), septo-optic dysplasia, tuberous sclerosis, and L1 syndrome.
Aicardi Syndrome
Definition
Aicardi syndrome is a neurodegenerative disorder first described in 1965, characterized by cerebral atrophy, intracerebral calcification of the basal ganglia, chronic cerebrospinal fluid lymphocytosis, and negative serologic investigations for common prenatal infections. It was classically characterized by agenesis of the corpus callosum, chorioretinal lacunae, and infantile spasms.
Incidence
More than 100 cases have been reported. It is seen only in females and 47,XXY males.
Etiology
The disorder is thought to be caused by an X-linked dominant de novo gene mutation with lethality in 46,XY males. The causative gene has not been identified.
Genetics
The genetics of Aicardi syndrome are unknown.
Diagnosis
The diagnosis is based on clinical features, including chorioretinal lacunae, brain MRI findings (corpus callosum dysgenesis, cerebral asymmetry, periventricular and intracortical gray matter heterotopia, choroid plexus cysts, choroid plexus papillomas, ventriculomegaly), and skeletal findings (abnormal vertebrae and missing ribs). Increased levels of interferon-α can been found in fetal blood and cerebrospinal fluid.
Associated Anomalies
Other features include characteristic facial features, GI difficulties, small hands, vascular malformations, skin pigmentary lesions, and increased incidence of tumors. Infrequently, there is associated cleft lip and palate.
Differential Diagnosis
Consider other causes of dysgenesis of the corpus callosum, including infectious causes.
Prognosis
Survival is highly variable, with mean age of death at about 8 years old. Patients have profound intellectual disability and severe global developmental delay. Medically refractory epilepsy with a variety of seizure types develops over time. Multiple antiepileptic drugs are generally required for seizure control. Costovertebral defects can lead to scoliosis. Constipation and other GI problems are frequent.
Recurrence Risk
The recurrence risk is less than 1%.

Meckel Syndrome
Definition
Meckel syndrome (MKS) is a lethal ciliopathy characterized by occipital encephalocele, postaxial polydactyly of the hands and feet, and renal cystic dysplasia. It is commonly associated with ductal plate malformations of the liver.
Synonyms
Dysencephalia splanchnocystica and Meckel-Gruber syndrome are synonyms for MKS.
Incidence
The prevalence of MKS in Finland is estimated to be 1 in 9000. In U.S. and European populations estimates are 1 in 13,250 to 1 in 140,000. MKS is the most common syndromic form of neural tube defects and polydactyly. MKS includes approximately 5% of all neural tube defects.
Etiology
MKS is a ciliopathy, and the proteins encoded by the genes implicated in the disorder encode for proteins involved in primary ciliary function.
Genetics
The disorder is genetically heterogeneous. The earliest implicated genes include MKS1 and MKS3 . Polydactyly is commonly found with MKS1 mutations and is rare with MKS3 mutations. Milder CNS phenotypes are seen with MKS3 mutations. The list of involved genes now includes at least 13 genes, MKS1 through 10 , TMEM231 , TMEM237 , and C5orf42 . Inheritance is autosomal recessive with significant phenotypic variability. Many of the same genes are also involved in Joubert syndrome.
Recurrence Risk
There is a 25% recurrence risk for MKS.
Diagnosis
The disorder can be detected prenatally as early as 11 to 14 weeks. Cystic dysplastic kidneys are seen in almost all cases of MKS (95-100%). The kidneys initially develop microscopic cysts that destroy the parenchyma and enlarge the organ up to 10 or 20 times. Occipital cephalocele is present in 60% to 80%. Maternal serum or amniotic fluid alpha-fetoprotein (AFP) level may be normal, as a membrane may cover the cephalocele. Other CNS findings include Dandy-Walker malformation and hydrocephalus. Postaxial polydactyly is present in 55% to 75% of fetuses. Other limb anomalies, such as bowing and shortening, may also be present. Liver histologic examination commonly demonstrates ductal plate malformations. Unfortunately, the first sonographic finding is often oligohydramnios, which makes the diagnosis more difficult to establish. Oligohydramnios is caused by renal dysfunction and develops early in the second trimester when the kidneys replace extracellular diffusion as the main source of amniotic fluid. Some cases of MKS have normal amniotic fluid; thus, the presence of normal fluid does not exclude the diagnosis.
Differential Diagnosis
Conditions that can present with similar findings include trisomy 13 and 18, Joubert syndrome, Bardet-Biedl syndrome, and Smith-Lemli-Opitz syndrome. Karyotype and molecular genetic testing can assist in clarifying the diagnosis.
Prognosis
MKS is a lethal disorder. Most infants are stillborn or die hours or days after birth. A few have survived to a few months of age.




Lissencephaly
Definition
Lissencephaly is a cerebral developmental disorder, with agyria of the brain, which may be accompanied by pachygyria, minimal or no hydrocephalus, a wide cortical mantle, and characteristic dysmorphic features. The reduced or absent brain gyri are caused by disturbed neuronal migration in the neocortex. Miller in 1963 and Dieker in 1969 provided the first descriptions.
Synonyms
See later discussion of specific syndromes.
Incidence
Incidence is uncertain, but estimates range from 11.7 to 40 cases per 1 million births.
Etiology
Many of the causative genes for lissencephaly have now been identified (see later).
Pathogenesis
Lissencephaly is due to abnormal cortical development in which migration of neurons from the ventricular zone (a region close to the lateral ventricles) have slow or arrested migration to the cortical plate, leading to reduced folding or stranded neurons. Lissencephaly is now classified into types including the following.
Classic Types.
Classic type lissencephaly is characterized by abnormally thick, four-layer cerebral cortex without other major brain anomalies. It is caused by mutations in 4 genes: PAFA, H1B1 (LIS1), DCX, and TUBA1A .
Miller-Dieker Syndrome.
Miller-Dieker syndrome (classic lissencephaly plus) is characterized by severe, classic lissencephaly, as well as facial dysmorphism (high forehead, micrognathia, short nose with anteverted nares, protuberant upper lip, bitemporal narrowing). Severe developmental delay and intellectual disability are also present. Other associated anomalies include omphalocele, cleft palate, and genital anomalies. Miller-Dieker syndrome results from chromosomal deletions at 17p13.3.
Cobblestone Cortical Malformation.
Cobblestone cortical malformation was previously known as type 2 lissencephaly and is distinct from classic lissencephaly. The cortex appears irregular or pebbled and is thinner. There may also be irregularity in the gray-white matter boundary, dilated ventricles, white matter abnormalities, brainstem hypoplasia, and cerebellar abnormalities. Although this group is genetically heterogeneous, it is most commonly due to defects in α-dystroglycan O -glycosylation. Known causative genes include POMT1, POMT2, POMGNT1, FKRP, FKTN, ISPD, and LARGE . The three associated phenotypes are Walker-Warburg syndrome, muscle-eye-brain disease, and Fukuyama congenital muscular dystrophy (ranging from most to least severe).
Walker-Warburg Syndrome.
Walker-Warburg syndrome is a congenital muscular dystrophy characterized by eye anomalies and cerebral malformations. A wide variety of eye and cerebral findings have been reported including: microphthalmia, buphthalmos, congenital glaucoma, cataract, optic nerve hypoplasia, persistent hyaloid artery, Dandy-Walker malformation, hydrocephalus, cephalocele, microcephaly, and agenesis of the corpus callosum. Most affected newborns die within the first year of life. No psychomotor development occurs.
Muscle-Eye-Brain Disease.
Muscle-eye-brain disease is a milder congenital muscular dystrophy in which ambulation may be acquired. Eye findings are common. The cerebral cortex demonstrates frontoparietal pachygyria, the cerebellar vermis is hypoplastic, and the brainstem is usually hypoplastic.
Fukuyama Congenital Muscular Dystrophy.
Fukuyama congenital muscular dystrophy is the most mild of the three phenotypes. Ambulation is often acquired. Eye findings are variable and often mild. Brain findings are variable, but typically less severe than the other phenotypes.
X-Linked Lissencephaly With Ambiguous Genitalia.
Males affected with X-linked lissencephaly have severe developmental delay, small or ambiguous genitalia, and seizures. Microcephaly, feeding difficulties, and growth failure are also present. Death in the first year is common. It is caused by muations in the gene ARX .
Baraitser-Winter Syndrome.
Baraitser-Winter syndrome is a rare syndrome with anterior-predominant pachygyria and characteristic facial features (hypertelorism, broad nose, ptosis, ridged metopic suture, arched eyebrows). Other common features include iris or retinal coloboma, sensorineural deafness, microcephaly, polyhydramnios, increased nuchal translucency, congenital heart defects, and renal tract anomalies. Intellectual disability and epilepsy are common. Caused by gain-of-function mutations in the genes ACTB and ACTG1 .
Lissencephaly With Cerebellar Hypoplasia.
Lissencephaly with anterior-predominant pachygyria and severe abnormalities of the cerebellum, brainstem, and hippocampus is caused by mutations in TUBA1A and RELN (Norman-Roberts syndrome).
Microlissencephaly.
Microlissencephaly is lissencephaly with head circumference at birth of less than three standard deviations. It is caused by mutations in the NDE1 gene.
Neu-Laxova Syndrome.
NLS is a lethal lissencephaly disorder inherited in an autosomal-recessive manner, characterized by growth restriction, microcephaly, agenesis of the corpus callosum, cerebellar hypoplasia, facial dysmorphism, hydrops, ichthyosis, extremity contractures, and syndactyly. It is due to an inborn error of serine metabolism, with causative mutations in the gene PHGDH .
Recurrence Risk
Recurrence risk is dependent on the particular lissencephaly syndrome.
Diagnosis
Sonographic diagnosis is difficult prior to the late second trimester, when the characteristic cerebral anomalies can be noted. The progressive microcephaly and failure of development of both sulci and gyri (which normally become well defined by 26 to 28 weeks) are suggestive of lissencephaly. Specific lissencephaly-related syndromes are difficult to differentiate prenatally. First-line genetic investigations involve chromosomal microarray, followed by next-generation sequencing multigene panels or exome sequencing.
Prognosis
Prognosis varies depending on the particular disorder.
Septo-Optic Dysplasia
Definition
Septo-optic dysplasia is a syndrome characterized by anomalies of cerebral midline structures, such as absence of the septum pellucidum, congenital optic nerve dysplasia, and panhypopituitarism, leading to multiple endocrine defects (diabetes insipidus, hypogonadotropic hypogonadism, hypothyroidism, adrenal insufficiency, abnormal thyrotropin-releasing hormone test, gonadotropin-releasing hormone test, and gonadotropin hormone–releasing hormone test). Septo-optic dysplasia may represent a mild form of holoprosencephaly.
Synonym
Septo-optic dysplasia is also known as de Morsier syndrome.
Incidence
The incidence is estimated at 1 in 10,000 newborns.
Etiopathology
Environmental factors such as viral infections, medications, and vascular disruption have been postulated to play a role. The implicated genes (see later) are involved in embryonic development and are essential for formation of the eyes, the pituitary gland, and forebrain structures such as the optic nerves.
Genetics
Mutations in HESX1, OTX2, and SOX2 are thought to be causative in a small subset of patients.
Recurrence Risk
The recurrence risk is unknown, depending on the cause.
Clinical Findings
- •
CNS: seizures, mental retardation, atrophy of the optic nerve, dilatation of the suprasellar cistern, empty sella, cortical atrophy and dystrophic corpus callosum, and anterior cephalocele.
- •
Face: hypotelorism, microphthalmia, visual impairment with nystagmus, unilateral or bilateral optic disk hypoplasia with double rim appearance (choroidal pigment in the outer margin and pale nerve tissue in the inner), variable visual loss, coloboma, strabismus, astigmatism, bilateral cleft lip and palate, high arched palate, and flat nasal bridge.
- •
Endocrine system: low growth rate and short stature. The most common problem is growth hormone deficiency (93%), followed by adrenocorticotropic hormone (ACTH) deficiency (57%), hypothyroidism (53%), diabetes, and gonadotropin deficiency. Hypothalamic dysfunction is the basic origin of these endocrine abnormalities. Septo-optic dysplasia is responsible for approximately 4% of all growth hormone deficiencies in children.
Diagnosis
Absence of the cavum septum pellucidum (CSP) is the most common finding. Sometimes, the standard sonographic views of the brain, obtained along the axial planes at midgestation, will fail to identify this absence. Absence of the CSP may not be detected because of the close proximity of the walls of the lateral ventricles, which are typically normal in size and may generate an artifact resembling a normal CSP. MRI is helpful for demonstration of hypoplastic optic tracts, allowing more definitive diagnosis. Hypotelorism, enlarged cerebral ventricles, communicating lateral ventricles, and bilateral cleft lip and palate have also been recognized prenatally.
Differential Diagnosis
Several variants with associated schizencephaly, dysgenesis of the corpus callosum, or microphthalmos, as well as incomplete forms, have been described. Lobar holoprosencephaly may also resemble septo-optic dysplasia.
Prognosis
The variable degree of intellectual disability (from minimal to severe), as well as the presence of multiple endocrine dysfunctions, affects the prognosis for each infant. Hyperthermia (in case of fever), dehydration (fever and diabetes insipidus), and other endocrine dysfunctions should be investigated and corrected.

Tuberous Sclerosis
Definition
Tuberous sclerosis is characterized by abnormalities of the skin (facial angiofibromas, hypochromic patches), brain (cortical tubers, subependymal nodules and giant cell astrocytomas, seizures, intellectual disability), kidneys (cysts, renal cell carcinomas, angiomyolipomas), heart (rhabdomyomas, arrhythmias), and lungs (lymphangioleiomyomatosis).
Synonyms
Tuberous sclerosis is also called Bourneville sclerosis and Bourneville disease.
Incidence
The incidence of tuberous sclerosis is up to 1 in 5800 births. Onset occurs in the first decade of life.
Etiology
Tuberous sclerosis is transmitted through autosomal-dominant inheritance. Two thirds of affected individuals have a de novo mutation. The expression is highly variable.
Pathogenesis
Tubers are the expression of an early disorder of embryogenesis. The greater the number of tubers, the greater the number of neurologic impairments. They can be found throughout the cerebral hemispheres, including in the subependymal region located in the walls of lateral ventricles and on the surface of the basal ganglia. They may extend into the ventricles including in the foramen of Monro area and can cause obstruction and hydrocephalus. They may also appear in the cortical gyri and sulcus terminalis. Tuberous sclerosis induces a reduction of the number of neurons, which are substituted by “monster” multinucleated giant neurons. The overgrowth of the fibrillary astrocytes can result in malignant astrocytomas. The sclerosis induces a demyelination, as well as calcium deposition in the glia and the blood vessels, which go through hyaline degeneration.
Clinical Findings
Tuberous sclerosis can present with a wide variety of signs that involve many organs as a consequence of the multifactorial origin of this genetic disorder ( Table 16-1 ).
| Classic triad (<50% of cases) |
| Facial lesions |
| Convulsions |
| Mental retardation |
| Central nervous system |
| Cortical hamartomas |
| Lesion of white substance * |
| Subependymal hamartomas (95%) |
| Typical localization: alongside lateral ventricle walls |
| Astrocytoma of subependymal giant cells * |
| Localization: foramen of Monro |
| Obstructive hydrocephaly |
| Kidney |
| Cysts |
| Angiolipoma |
| Cardiovascular |
| Rhabdomyoma |
| Aneurysm, stenosis, and vascular ectopia |
| Liver |
| Leiomyoma |
| Adenoma |
Diagnosis
The diagnosis is usually suggested by the discovery of cardiac tumors, which resemble small uterine myomas (round, usually well-delineated homogeneous masses). Between 51% and 86% of cardiac rhabdomyomas are seen in patients with tuberous sclerosis. Occasionally, the finding of a rhabdomyoma during routine second trimester ultrasound examination may lead to the recognition that the mother is affected. Cardiac rhabdomyomas increase prenatally, may regress in early infancy, remain unchanged during childhood, and regress in adolescence. The rhabdomyomas may cause rhythm disruptions (Wolff-Parkinson-White syndrome, supraventricular tachycardia, paroxysmal arrhythmias), as well as obstructions or regurgitation. Renal angiofibromas have not been recognized prenatally, although this may simply be a matter of time. Some recent unpublished reports have demonstrated that the periventricular subependymal nodules may also be detected prenatally. 2D Doppler echocardiography is a useful, noninvasive method to diagnose fetal cardiac rhabdomyomas and to monitor their influence on fetal cardiac function. However, it does not help to determine which fetuses affected by rhabdomyomas have tuberous sclerosis. Some studies report that 39% of prenatal suspected cardiac rhabdomyomas will be diagnosed at birth as a tuberous sclerosis syndrome. Family history remains the strongest predictor of the syndrome in prenatal counseling, whereas size of the cardiac tumor cannot reliably be used. The presence of more than one rhabdomyoma is more likely to be associated with tuberous sclerosis than a single identified lesion. MRI can be performed to assess for the associated malformations.
Genetics
Tuberous sclerosis is caused by defects or mutations in one of two genes, TSC1 and TSC2 . TSC1 produces a protein called hamartin. TSC2 produces a protein called tuberin. These proteins act as tumor suppressors, which are agents that regulate cell proliferation and differentiation. At least 1% of patients with tuberous sclerosis have somatic mosaicism for TSC1 or TSC2 .
Differential Diagnosis
The predominant prenatal finding is rhabdomyomas. Other cardiac tumors, such as a fibroma, should also be considered.
Recurrence Risk
Recurrence risk is generally low in cases of new mutations, but up to 1% to 2% due to gonadal mosaicism, 50% if a parent is affected.
Prognosis
As long as hydrops is not present from the presence of the rhabdomyomas, the prognosis depends on other complications of the disorder. CNS tumors are the leading cause of morbidity and fatality. Renal disease is the second leading cause of early death. Because of the wide variability of expression, an accurate prediction of a child’s phenotype is difficult to infer from the status of the parent. Genetic evidence indicates that the degree of intelletual disability depends on the particular genetic alteration present.



L1 Syndrome
Definition
L1 syndrome includes a spectrum of phenotypes (see following synonyms) and is generally characterized by severe hydrocephalus, adducted thumbs, spasticity, and severe intellectual disability.
Synonyms
Synonyms for L1 syndrome include X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS), MASA (mental retardation, aphasia, spastic paraplegia, adducted thumbs) syndrome, SPG1 (X-linked complicated hereditary spastic paraplegia type 1), and X-linked corpus callosum agenesis.
Incidence
The incidence is 1 in 30,000 births, accounting for 5% to 10% of males with congenital hydrocephalus not associated with another syndrome.
Pathogenesis
Mutations in the L1CAM gene are thought to lead to the clinical phenotype. L1CAM is an adhesion surface protein involved in transmembrane signaling and is essential for the development and function of neurons.
Diagnosis
Clinical findings include hydrocephalus with or without stenosis of the aqueduct of Sylvius, as well as corpus callosum agenesis or dysgensis, cerebellar hypoplasia, small brainstem, and bilateral absence of the pyramids. Bilateral absence of the pyramids on MRI or at autopsy is pathognomonic. Prenatally, hydrocephalus can be seen, but is often not seen prior to 20 to 24 weeks’ gestation, and sometimes is not seen even in the third trimester of pregnancy.
Genetics
L1 syndrome is caused by mutations in L1CAM gene. Inheritance is X-linked recessive.
Recurrence Risk
Recurrence risk is 50% for women who are carriers of an L1CAM mutation.
Associated Anomalies
Hirschsprung disease has been seen in some individuals with L1 syndrome.
Differential Diagnosis
Other syndromic and nonsyndromic causes of hydrocephalus should be excluded.
Prognosis
The phenotype can range from mild to severe even within the same family.
Management
A multidisciplinary team is required for optimal management. Head imaging should be performed. Surgical treatment is often required to relieve hydrocephalus. Regular neurologic, developmental evaluation and follow-up are needed. Surgery for adducted thumbs is generally not indicated.
Cranium/Face
Craniosynostoses
Craniosynostosis has been reported in over 150 genetic disorders and is found in 1 per 2000 to 2500 live births. Many of the craniosynostoses are caused by mutations in one of the FGFR genes, including Apert syndrome, Beare-Stevenson syndrome, Crouzon syndrome, Crouzon syndrome with acanthosis nigricans, FGFR2 -related isolated coronal synostosis, Jackson-Weiss syndrome, Muenke syndrome, and Pfeiffer syndrome. Although Muenke syndrome and FGFR2 -related isolated coronal synostosis are characterized by coronal craniosynostosis only, the others have related facial features and extremity findings. FGFR -related craniosynostoses have been associated with advanced paternal age. Syndromic craniosynostoses involving other genes include Antley-Bixler syndrome (caused by mutations in the POR gene), Carpenter syndrome (caused by mutations in RAB23 ), and Saethre-Chotzen syndrome (caused by mutations in TWIST1 ).
In general, management of the craniosynostoses should include involvement of a multidisciplinary craniofacial clinic at a pediatric medical center, as a series of staged surgical procedures is often required for treatment and generally involves craniotomy and fronto-orbital advancement. Surgical correction of limb defects may also be needed, depending on the particular anomalies. Early treatment can decrease the risk of secondary complications, such as hydrocephalus and intellectual disability. If proptosis is severe, ophthalmologic lubrication can prevent exposure keratopathy. The prognosis for each affected individual is most dependent on the particular anomalies present and less on the specific craniosynostosis syndrome.
A few of the more common disorders are discussed in more detail above ( Table 16-2 ).
| Disorder | Thumbs | Hands | Great Toes | Feet | Genetic Basis |
|---|---|---|---|---|---|
| Crouzon syndrome | Normal | Normal | Normal | Normal | FGFR2 |
| Crouzon syndrome with acanthosis nigricans (AN) | Normal | Normal | Normal | Normal | FGFR3 |
| Apert syndrome | Occasionally fused to fingers | Soft tissue ± bone syndactyly | Occasionally fused to toes | Soft tissue ± bone syndactyly | FGFR2 |
| Pfeiffer syndrome | Broad, medially deviated | Variable brachydactyly | Broad, medially deviated | Variable brachydactyly | FGFR1 (5% of type 1); FGFR2 (most) |
| Muenke syndrome | Normal | ± Carpal fusion | ± Broad | ± Tarsal fusion | FGFR3 |
| Jackson-Weiss syndrome | Normal | Variable | Broad, medially deviated | Abnormal tarsals | FGFR2 |
| Beare-Stevenson syndrome | Normal | Normal | Normal | Normal | < FGFR2 |
| FGFR2 -related isolated coronal synostosis | Normal | Normal | Normal | Normal | FGFR2 |
Antley-Bixler Syndrome
Definition.
Antley-Bixler syndrome is a sterol biosynthesis disorder characterized by craniosynostosis of the coronal and lambdoidal sutures, brachycephaly with frontal bossing, and facial dysmorphism (proptosis, downslanting palpebral fissues, severe depression of the nasal bridge, choanal stenosis or atresia, small mouth, high-arched narrow palate, low-set protruding ears). Hydrocephalus can be present. Limb anomalies include radiohumeral synostosis, bowing of the ulnas and femurs, slender hands and feet, contractions of the proximal interphalangeal joints, advanced bone age, and fractures.
Synonym.
Antley-Bixler syndrome is also known as trapezoidocephaly-multiple synostosis syndrome.
Etiology.
Caused by cytochrome P450 oxidoreductase (POR) deficiency, Antley-Bixler syndrome is the most severe end of the phenotypic spectrum of POR deficiency.
Incidence.
The incidence is unknown. Since POR mutations were first reported in 2004, approximately 50 affected individuals have been reported. More mildly affected individuals are likely underreported.
Pathogenesis.
The mechanism by which POR deficiency leads to multiple malformations remains under investigation. Ambiguous genitalia result from disordered steroidogenesis. Recently, cytochrome P450 activity in bone has been implicated in normal bone development, and, thus, POR deficiency may disrupt proper bone formation.
Diagnosis.
Malformations that may be detected on prenatal ultrasound examination include abnormal skull shape, facial dysmorphism, skeletal abnormalities (bowed femora, bilateral radioulnar synostosis), and ambiguous genitalia. During pregnancy, low maternal estriol may be found on serum screening. Maternal urine may also have increased fetal steroids, including the pregnenolone metabolite epiallopregnanediol and the androgen metabolite androsterone. POR deficiency can be diagnosed in affected infants by the detection of urinary sterol or steroid abnormalities, including increased pregnenolone and progesterone metabolites and an elevated ratio of metabolites associated with deficiency of 17-hydroxylase and 21-hydroxylase. Serum concentrations of pregnenolone, progesterone, 17-OH pregnenolone, and 17-OH progesterone may be elevated at baseline or following ACTH stimulation. Some cases are found on newborn screening (NBS), but NBS is not sensitive enough to detect all cases.
Genetics.
Antley-Bixler syndrome is caused by mutations in the gene encoding cytochrome P450 reductase ( POR ). Inheritance is autosomal recessive.
Recurrence Risk.
Recurrence risk is 25%.
Associated Anomalies.
Congenital heart disease, renal anomalies, abnormalities of the female genitalia, and signs of congenital adrenal hyperplasia are also present. Some degree of intellectual disability is often seen, but intelligence can also be normal.
Differential Diagnosis.
Other forms of congenital adrenal hyperplasia as well as other craniosynostosis syndromes should be considered.
Prognosis.
Prognosis improves with age. In infancy, respiratory complications can lead to early death.
Management.
Multidisciplinary evaluation is needed. Airway management is a primary concern owing to choanal atresisa/stenosis. Functional adrenal studies should be performed and hydrocortisone replacement therapy initiated if low cortisol levels are found. Genital abnormalities are often treatable and should be addressed.
Apert Syndrome
Definition.
Apert syndrome is characterized by craniosynostosis, midface and orbital hypoplasia, and bilateral syndactyly of the hands and feet. These findings are accompanied by variable degrees of mental retardation in 50% of cases.
Incidence.
The incidence of Apert syndrome is 1 per 100,000 live births. It is seen in 4% to 5% of craniosynostosis cases.
Synonyms.
Apert syndrome is also known as acrocephalosyndactyly type 1 and Apert-Crouzon disease.
Etiology.
Apert syndrome has autosomal dominant inheritance. Most of the cases are sporadic, resulting from de novo mutations. The disorder is associated with advanced paternal age.
Sonographic Findings.
Brachycephaly and acrocephaly, high forehead, flat occiput, craniosynostosis involving the coronal sutures, flat face, and hypertelorism are seen. Other ultrasound findings include agenesis of the corpus callosum, mild ventriculomegaly, and fusion of the cervical vertebrae at the level of C5-C6. In the extremities, syndactyly of both bone and soft tissue is seen, involving the second, third, and fourth fingers. Polyhydramnios (caused by the decreased fetal swallowing) and increased nuchal translucency in the first trimester have also been reported.
Diagnosis.
Typical findings include bicoronal synostosis of the coronal sutures, flattened occiput, a wide, steep forehead, hypoplastic orbits with exophthalmos and hypertelorism, short nose with depressed nasal bridge, large ears, high palate (occassional cleft palate), and dental crowding. Hydrocephalus is rare. Hearing loss is common. Symmetric syndactyly of the limbs (“mitten-like” hands and feet) is present at least from the second to fourth digits in both the bony and soft tissue. Cardiovascular and genitourinary anomalies are present in 10% of patients.
Genetics.
Caused by mutations in the FGFR2 genes, most commonly substitutions S252W and P253R. Genetic molecular studies are recommended for the fetus (by chorionic villus sampling or amniocentesis) and for parents when Apert syndrome is suspected. Inheritance is autosomal dominant with complete penetrance.
Differential Diagnosis.
Other syndromes with craniosynostosis such as Crouzon, Pfeiffer, Carpenter, and Saethre-Chotzen syndromes should be considered. Molecular genetic studies can exclude these disorders. The pattern of syndactyly is the most helpful for the diagnosis of Apert syndrome.
Recurrence Risk.
Most mutations are de novo and the recurrence risk is low. If one of the parents carries the disorder, the recurrence risk is 50%.


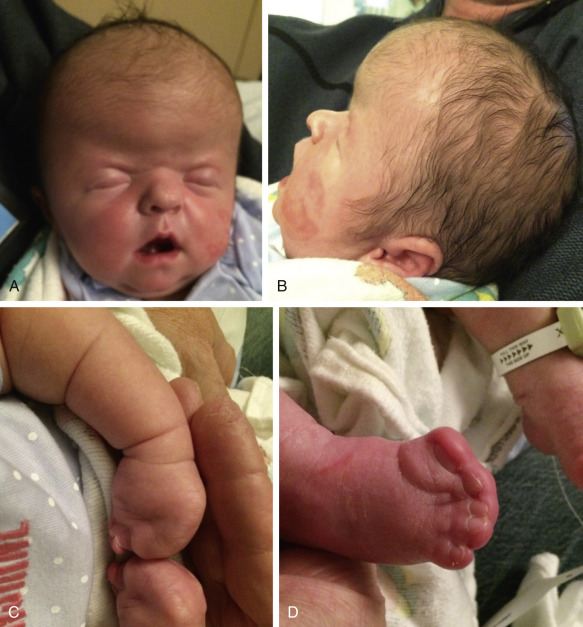
Carpenter Syndrome
Definition.
Carpenter syndrome is characterized by craniosynostosis with preaxial polydactyly of the feet. Hand anomalies include brachydactyly, syndactyly, and aplasia or hypoplasia of the middle phalanges.
Synonym.
Acrocephalopolysyndactyly type II is another name for Carpenter syndrome.
Incidence.
Carpenter syndrome is rare. Approximately 70 cases have been described.
Etiology/Pathogenesis.
RAB23 is a member of the RAB guanosine triphosphatase (GTPase) family of vesicle transport proteins and acts as a negative regulator of hedgehog signaling. Abnormal hedgehog signaling may lead to much of the clinically recognized phenotype.
Diagnosis.
Unlikely other syndromic craniosynostoses, in which coronal sutures are most frequently affected, in Carpenter syndrome, fusion of the midline sutures (metopic and sagittal) is common. In severe cases, this leads to cloverleaf skull. On prenatal ultrasound examination, reported features include cystic hygroma, abnormal skull shape, bowed femora, polydactyly, and complex heart defects.
Genetics.
Carpenter syndrome is caused by mutations in RAB23 . The inheritance is autosomal recessive. A subtype of Carpenter syndrome with lateralization defects is thought to be caused by mutations in MEGF8 .
Recurrence Risk.
Recurrence risk is 25%.
Associated Anomalies.
Obesity, umbilical hernia, hearing loss, cryptorchidism, and cardiac defects are often present. Craniosynostosis leads to distinctive facial features. The degree of intellectual disability varies.
Differential Diagnosis.
Features overlap with Greig cephalopolysyndactyly syndrome. Molecular genetic testing can clarify the diagnosis.
Prognosis.
Phenotype is extremely variable, even within the same family. Life expectancy is shortened.
Crouzon Syndrome
Definition.
Crouzon syndrome is an FGFR -related craniosynostosis characterized by premature fusion of the coronal and frontosphenoidal sutures leading to brachycephaly and a prominent forehead. Facial dysmorphism (proptosis from orbital and midface hypoplasia, external strabismus, mandicular prognathism) is present and extremities are normal.
Incidence.
The incidence of Crouzon syndrome is 1.6 per 100,000 live births. It is seen in 4.5% of craniosynostosis cases.
Diagnosis.
Sonographic findings include brachycephaly, midface hypoplasia, and a wide anterior cranial base. Hands and feet are normal. Exophthalmos is always present. About 20% of patients develop optic atrophy. Dental crowding and an open bite are common.
Genetics.
Caused by mutations in FGFR2 (except for Crouzon syndrome with acanthosis nigricans, which is caused by mutations in FGFR3 , seen in ~5% of individuals with Crouzon syndrome). Inheritance is autosomal dominant with complete penetrance and variable expressivity.
Recurrence Risk.
Recurrence risk is 50% if one parent carries the causative mutation. Occassionally it appears de novo.
Associated Anomalies.
Progressive hydrocephalus is seen in 30% of cases and can lead to tonsillar herniation. Sacrococcygeal tail can also be seen. Cardiovascular anomalies and cleft lip and palate have been reported but are rare.
Differential Diagnosis.
Other FGFR -related craniosynostoses should be considered, including Crouzon syndrome with acanthosis nigricans (should consider if choanal atresia is present).
Prognosis.
Intellect is generally normal, hearing loss is common, and complications related to hydrocephalus can be life threatening if not treated appropriately.
Pfeiffer Syndrome
Definition.
Pfeiffer syndrome is characterized by bilateral coronal craniosynostosis, midface hypoplasia, syndactyly of hands and feet, and congenitally enlarged thumbs. Conductive hearing loss is present in more than 80% of patients. The syndrome is divided in three clinical subtypes, with diagnostic and prognostic implications:
- •
Type I: classic appearance with craniosynostosis (leading to turribrachycephaly), broad thumbs, and syndactyly. This form is compatible with life, and patients often have normal intelligence.
- •
Type II: cloverleaf skull, ocular proptosis, broad thumbs, variable visceral anomalies, elbow ankylosis, choanal atresia, and CNS involvement (hydrocephalus). Intellectual disability is common, and this form usually results in early death.
- •
Type III: craniosynostosis, severe ocular proptosis without cloverleaf skull, elbow ankylosis, and variable visceral anomalies. Affected fetuses have severe neurologic compromise (including hydrocephalus and tonsillar herniation), with poor prognosis and early death.
Synonym.
Pfeiffer syndrome is also known as acrocephalosyndactyly type 5.
Incidence.
The incidence is 1 per 100,000 for all types combined.
Genetics.
Pfeiffer syndrome is genetically heterogeneous and is caused by mutations in either FGFR2 or FGRF1 (type I). Frequently, no mutation can be identified. Type I has autosomal dominant inheritance with complete penetrance and variable expressivity. Most mutations in type II and III are de novo and associated with advanced paternal age.
Recurrence Risk.
Recurrence risk is 50% if a parental mutation is present. If the mutation is de novo, recurrence risk is very low.
Diagnosis.
Sonographic signs of Pfeiffer syndrome include craniofacial anomalies (brachycephaly, acrocephaly, craniosynostosis of the coronal suture, hypertelorism, small nose, and low nasal bridge) and hands and feet anomalies (partial syndactyly of second and third fingers and second, third, and fourth toes, broad thumb, and big toe).
Associated Anomalies.
Choanal atresia, tracheomalacia and bronchomalacia, cloverleaf skull, fused vertebra, Arnold-Chiari malformation, hydrocephalus, and imperforate anus are associated with this syndrome. After birth, seizures and intellectual disability may be seen.
Differential Diagnosis.
Saethre-Chotzen and Jackson-Weiss syndromes should be considered in the differential diagnosis.
Prognosis.
The prognosis depends on the severity of associated anomalies, in particular the severity of the CNS compromise. Type I generally has a good prognosis. Types II and III are not compatible with life, and death occurs early.
Saethre-Chotzen Syndrome
Definition.
Saethre-Chotzen syndrome is characterized by coronal synostosis (unilateral or bilateral), facial asymmetry, ptosis, and characteristic ear appearance (small pinna with a prominent crus). Often present is 2-3 syndactyly of the hand.
Synonyms.
Synonyms include acrocephalosyndactyly type III and Robinow-Sorauf syndrome (a mild variant).
Incidence.
Incidence is 1 in 25,000 to 50,000 individuals.
Diagnosis.
A clinical diagnosis can be made based on craniosynostosis (usually of the coronal sutures), brachycephaly, low frontal hairline, ptosis, facial asymmetry, small ears, and limb anomalies (2-3 syndactyly of the hand, brachydactyly, hallux abnormalities).
Genetics.
Saethre-Chotzen syndrome is caused by mutations in TWIST1 ; sequencing and deletion/duplication analysis should be performed. Mutations have also been reported in FGFR2 . Inheritance is autosomal dominant with incomplete penetrance and variable expressivity.
Recurrence Risk.
If one parent also carries the causative mutation, recurrence risk is 50%. It is significantly less if the mutation is de novo.
Associated Anomalies.
Associated anomalies include short stature, parietal foramina, radioulnar synostosis, cleft palate, maxillary hypoplasia, hypertelorism, congenital heart disease, and segmentation defects of the vertebrae.
Differential Diagnosis.
Many features are shared with Muenke syndrome, and, thus, genetic evaluation should also include analysis of the FGFR2 and FGFR3 genes. Pfeiffer syndrome and Jackson-Weiss syndrome should also be considered.
Prognosis.
Intelligence is often normal, although mild to moderate intellectual and developmental delay has been reported. Conductive and sensorineural hearing loss may be present.
Facial Anomalies
Branchiooculofacial Syndrome
Definition.
Branchiooculofacial syndrome (BOFS) is characterized by branchial skin defects (thin skin, erythematous lesions, erosions), ocular anomlies (microphthalmia, anophthalmia, coloboma, nasolacrimal duct stenosis/atresia), and facial anomalies (hypertelorism, broad nasal tip, upslanted palpebral fissues, cleft lip ± cleft palate, upper lip pits, lower facial weakness). Ear anomalies are common.
Incidence.
BOFS is rare. Fewer than 100 well-described cases have been reported.
Diagnosis.
Diagnosis is made on the clinical constellation of branchial skin defect, ocular anomaly, and characteristic facial appearance.
Genetics.
BOFS is caused by mutations in the gene TFAP2A . Inheritance is autosomal dominant. De novo mutations are present in 50% to 60% of affected individuals. Penetrance is complete.
Recurrence Risk.
Recurrence risk is 50% if a parental mutation is found.
Associated Anomalies.
Other anomalies include thymic anomalies, renal anomalies, hypoplastic teeth, dysplastic nails, premature hair graying, and defects in hearing and vision.
Prognosis.
Intellect is generally normal. Feeding difficulties, as well as cosmetic, visual, hearing, and speech issues are often present.
Frontonasal Dysplasia
Definition.
Frontonasal dysplasia is characterized by a broad forehead, widow’s peak hairline, ocular hypertelorism, and abnormal nostrils (notched or completely divided) with absence of the nasal tip. A midline defect in the frontal bone (cranium bifidum) is also commonly seen.
Synonyms.
Synonyms include median cleft face syndrome, frontonasal malformation, and frontorhiny.
Incidence.
Incidence is unknown. Frontonasal dysplasia is rare; at least 100 cases have been reported in the literature.
Pathogenesis.
Frontonasal dysplasia is due to malformation of the frontonasal elevation. The implicated genes ( ALX1, ALX 3, and ALX 4 ) encode transcription factors that regulate development of the eyes, nose, and mouth.
Diagnosis.
The diagnosis is based on the constellation of eye, forehead, and nose findings described earlier.
Genetics.
The disorder is genetically heterogeneous, with autosomal recessive, autosomal dominant, and X-linked forms of inheritance all reported. The autosomal recessive form is associated with mutations in ALX1 and ALX3 . The autosomal dominant form is associated with ALX4 gene mutations.
Recurrence Risk.
The recurrence risk is dependent on mode of inheritance.
Prognosis.
Most individuals have normal intellectual development. Overall prognosis depends on the severity of the malformations and whether or not surgical intervention can improve breathing and feeding issues.
Management.
Multistage craniofacial surgery is generally needed; for less severe malformations, surgery is often performed at age 6 to 8 and can have excellent cosmetic results.
Goldenhar Syndrome
Definition.
Goldenhar syndrome is characterized by hemifacial microsomia, epibulbar dermoids, preauricular appendages, ear hypoplasia, transverse facial clefts, asymmetry of the skull, and spinal anomalies (vertebral segmentation errors).
Incidence.
Incidence is 1 in 3500 to 1 in 26,000 live births. Male-to-female ratio is 3 : 2.
Synonyms.
Synonyms include oculo-auriculo-vertebral dysplasia, hemifacial microsomia, craniofacial microsomia, Goldenhar-Gorlin’s syndrome, and facio-auriculo-vertebral dysplasia.
Etiology.
Goldenhar syndrome is caused by developmental abnormalities of the first and second branchial arches.
Diagnosis.
Findings are similar to Treacher Collins syndrome (see later) except that they are unilateral leading to significant facial asymmetry. Other features include dystopia, microtia, choanal atresia, cleft palate ± lip, jaw malocclusion, vertebral anomalies, cardiac anomalies, genitourinary anomalies, and CNS involvement.
Pathogenesis.
Possible fetal hemorrhage in the region of the first and second branchial arches when the blood supply of these arches switches from the stapedial artery to the external carotid artery.
Genetics.
Genetics for Goldenhar syndrome are unknown. Most cases are thought to be sporadic, although both autosomal recessive and autosomal dominant inheritance have been reported. Transmission is linked in some cases to a region on chromosome 14q32.
Differential Diagnosis.
Treacher Collins syndrome, Kaufman syndrome (oculocerebrofacial syndrome), acrofacial dysostosis, CHARGE syndrome ( c oloboma of the eye, h eart defects, a tresia of the choanae, r etarded mental and growth development, g enital anomalies, and e ar anomalies), VACTERL, and Nager syndrome should be considered.
Prognosis.
Treatment is generally cosmetic with multiple reconstructions required in some cases. Airway and vertebral complications have been reported.
Recurrence Risk.
Recurrence risk is minimal unless an autosomal dominant or autosomal recessive inheritance pattern is suggested.
Management.
Malar and orbital reconstructions are generally needed and are often performed after age 6. Structural anomalies of the eyes and ears can be corrected with plastic surgery.
Gorlin Syndrome
Definition.
Gorlin syndrome is characterized by the development of jaw keratocysts and multiple basal cell carcinomas, as well as a characteristic facial appearance with macrocephaly, prominent forehead, coarse facial features, and facial milia.
Synonym.
Gorlin syndrome has also been referred to as nevoid basal cell carcinoma syndrome (NBCCS).
Incidence.
Incidence is estimated to be as high as 1 in 18,976.
Pathogenesis.
Mutations in PTCH1 lead to alterations in the hedgehog signaling pathway leading to neoplastic cell growth.
Diagnosis.
Prenatally, cranial and cerebral malformations can be seen, including macrocephaly and ventriculomegaly. Diagnosis is based on major clinical criteria including calcification of the falx, jaw keratocyst, palmar/plantar pits, and multiple basal cell carcinomas, as well as minor criteria.
Genetics.
Gorlin syndrome results from mutations in the gene PTCH1 . Inheritance is autosomal dominant. From 20% to 30% of mutations are de novo. Penetrance is complete; expressivity can be variable.
Recurrence Risk.
If a parental mutation is present, recurrence risk is 50%.
Associated Anomalies.
Most individuals have skeletal anomalies (bifid ribs, wedge-shaped vertebrae) and ectopic calcification (in the falx). Cardiac and ovarian fibromas can be seen. Children can develop medulloblastoma (5%). Other anomalies include lymphomesenteric or pleural cysts, cleft lip/palate, polydactyly, and ocular anomalies.
Differential Diagnosis.
When macrocephaly is detected prenatally, the differential diagnosis includes several overgrowth syndromes, such as Sotos syndrome and Beckwith-Wiedemann syndrome (BWS).
Prognosis.
Life expectancy is not changed. Cosmetic issues with multiple skin tumors and their removal can lead to social difficulties and can affect quality of life.
Management/Prevention.
Radiotherapy can lead to the development of thousands of basal cell carcinomas and should be avoided if possible. X-ray exposure should be limited. Direct sun exposure should be avoided.
Hallerman-Streiff Syndrome
Definition.
Hallerman-Streiff syndrome is characterized by brachycephaly with frontal bossing, micrognathia, beaked nose, eye anomalies (microphthalmia, cataracts), dental anomalies, hypotrichosis, skin atrophy (especially of the face), and short stature.
Incidence.
The syndrome is rare, with approximately 150 cases reported.
Diagnosis.
Diagnosis is based on the clinical criteria described earlier.
Genetics.
Generally thought to be sporadic, in some cases, the syndrome has been associated with mutations in GJA1 with autosomal recessive inheritance.
Recurrence Risk.
Recurrence risk is unknown, but thought to be low.
Differential Diagnosis.
Many features overlap with oculodentodigital dysplasia (ODDD) and other progeroid syndromes.
Prognosis.
Intellectual disability is present in approximately 15%. Upper airway obstruction can be significant and can lead to cardiac failure and early death.
Management.
Initial management is focused on airway issues; later management includes cosmetic surgical procedures.
Nager Syndrome
Definition.
Nager syndrome is characterized by preaxial limb anomalies (radial hypoplasia/absence, thumb hypoplasia/absence, triphalangeal thumbs, radioulnar synostosis) and facial anomalies (malar hypoplasia, downward slanting palpebral fissures, lower eyelid coloboma, severe micrognathia).
Synonym.
Nager syndrome is also known as preaxial acrofacial dysostosis.
Incidence.
Incidence is unknown, but the syndrome is rare.
Diagnosis.
Diagnosis is based on the clinical criteria described earlier, including craniofacial and limb malformations. Prenatally, severe micrognathia and upper limb anomalies may be seen.
Genetics.
The genetic basis is heterogeneous. Recent reports suggest that haploinsufficiency of the gene SF3B4 leads to the phenotype in families with suggested autosomal dominant inheritance, as well as many individuals with de novo mutations. Autosomal recessive inheritance has also been suggested in some cases.
Recurrence Risk.
Recurrence risk depends on mode of inheritance.
Differential Diagnosis.
Facial anomalies are similar to those seen with Treacher Collins syndrome. Differential diagnosis should include Miller syndrome.
Prognosis.
Upper airway issues can be significant, requiring tracheostomy. Hearing loss, vision loss, and hand anomalies can affect development and quality of life
Oral-Facial-Digital Syndrome, Type I
Definition.
Oral-facial-digital syndrome, type I (OFD1), is characterized by anomalies in multiple areas. including the mouth (lobed tongue, tongue lesions, cleft palate, dental anomalies), face (hypertelorism, alae nasi hypoplasia, median cleft, micrognathia), digits (brachydactyly, syndactyly, polydacyly of the hands, duplicated great toe), brain (intracerebral cysts, cerebellar agenesis, agenesis of the corpus callosum), and kidneys (polycystic kidneys).
Synonym.
OFD1 is also known as Papillon-Léage-Psaume syndrome.
Incidence.
Almost all affected individuals are female, although males with OFD1 have also been described.
Pathogenesis.
OFD1 is due to dysfunction of primary cilia.
Diagnosis.
Many individuals are diagnosed at birth based on the characteristic anomalies. Some cases are diagnosed after polycystic kidney disease is found.
Genetics.
The syndrome is due to mutations in the OFD1 gene. Inheritance is X-linked dominant. Approximately 75% of individuals have no family history of OFD1. Penetrance is high, with highly variable expressivity.
Recurrence Risk.
Recurrence risk is 50% in pregnancies of a female with OFD1. The risk is low if no maternal mutation is present.
Differential Diagnosis.
Differential diagnosis includes the other oral-facial-digital syndromes, types 2 to 9, as well as other disorders with cystic renal disease, and Meckel-Gruber syndrome.
Prognosis.
Up to 50% of individuals have intellectual disability, but generally mild. Seizure disorder can be present.
Management.
Surgery for oral and facial anomalies is often needed. Renal disease should be monitored.
Pierre Robin Sequence
Definition.
The Pierre Robin sequence consists of the occurrence of three anomalies together: cleft palate, micrognathia, and glossoptosis. These anomalies are found together in multiple syndromes.
Synonyms.
Pierre Robin sequence is also called cleft palate-micrognathia-glossoptosis, Pierre Robin syndrome, and Robin anomalad.
Incidence.
Incidence is 1 in 8500 to 1 in 14,000 births. One half of patients with Pierre Robin sequence have an underlying syndrome.
Etiology.
Inheritance is autosomal-recessive, with a few X-linked cases. Some authors suggest a prenatal and neonatal brainstem dysfunction as a neuroembryologic hypothesis to explain the onset of some cases of Pierre Robin sequence.
Diagnosis.
Micrognathia is most visible on prenatal ultrasound imaging. Isolated cleft palate is more difficult to detect prenatally. Polyhydramnios may be seen as a result of swallowing difficulty.
Genetics.
This disorder is heterogeneous; it can be monogenic, chromosomal, or related to teratogens. The cause is unknown in some cases. Multiple forms of inheritance are involved, including autosomal dominant, autosomal recessive, and X-linked.
Differential Diagnosis.
Differential diagnosis includes syndromes that involve Pierre Robin sequence, such as Stickler syndrome, as well as fetal alcohol syndrome.
Associated Anomalies.
Other anomalies depend on the exact diagnosis. Cardiac abnormalities are common with an incidence approaching 20%.
Prognosis.
Upper airway obstruction, neonatal respiratory distress, and feeding problems are common issues.
Associated Syndromes.
See Table 16-3 for a complete list of associated syndromes.
| Condition | OMIM | Causative Gene | Inheritance Pattern | Additional Features |
|---|---|---|---|---|
| Primary Skeletal Dysplasias | ||||
| Campomelic dysplasia | 114290 | SOX9 | Autosomal dominant | Short stature, campomelia, hearing loss, scoliosis |
| Stickler syndrome | 108300, 604841, 184840 | COL2A1, COL11A1, COL11A2 | Autosomal dominant | Myopia, retinal detachment, hearing loss, mild short stature |
| Spondyloepiphyseal dysplasia congenita | 183900 | COL2A1 | Autosomal dominant | Short spine, pectus carinatum, myopia, normal hands and feet |
| Kniest dysplasia | 156550 | COL2A1 | Autosomal dominant | Short trunk, kyphosis, lumbar lordosis, short limbs, reduced joint mobility |
| Marshall syndrome | 154780 | COL11A1 | Autosomal dominant | Hypoplastic nasal bones, congenital cataracts, myopia, sensorineural hearing loss |
| Otospondylomegaepiphyseal dysplasia | 277610, 215150 | COL11A2 | Autosomal dominant | Sensorineural hearing loss, enlarged painful joints, short stature, short limbs |
| Diastrophic dysplasia | 222600 | SLC26A2 | Autosomal recessive | Short stature, short limbs, joint contractures, talipes equinovarus, kyphoscoliosis |
| Osteopathia striata with cranial sclerosis | 300373 | WTX | X-linked dominant | Macrocephaly, frontal bossing, hearing loss, skull base sclerosis, linear metaphyseal bands |
| Otopalatodigital syndrome type II | 304120 | FLNA | X-linked semidominant | Frontal bossing, short thumbs and halluces, bowed long bones, conductive hearing loss |
| Multiple Congenital Anomaly Syndromes | ||||
| Catel-Manzke syndrome | 302380 | Unknown | Probably autosomal recessive | Clinodactyly of index finger due to accessory ossicle between metacarpal and proximal phalanx, congenital cardiac defects |
| Toriello-Carey syndrome | 217980 | Unknown | Possibly autosomal recessive | Agenesis corpus callosum, telecanthus, developmental delay |
| Treacher Collins syndrome | 154500 | TCOF1 | Autosomal dominant | Downslanting palpebral fissures, lower eyelid colobomas, zygomatic hypoplasia |
| Nager preaxial acrofacial dysostosis | 154400 | SF3B4 | Autosomal dominant | Downslating palpebral fissures, absence of medial third lower lid lashes, conductive hearing loss, radial ray defects |
| Miller postaxial acrofacial dysostosis | 263750 | DHODH | Autosomal recessive | Lower lid ectropion, cupped ears, hearing loss, bilateral absence of fifth fingers, supernumerary nipples |
| Mandibulofacial dysostosis Guion-Almeida type | 610536 | EFTUD2 | Autosomal dominant | Microcephaly, developmental delay, dysplastic ears, preauricular tags, choanal atresia |
| Auriculocondylar syndrome | 602483, 614669 | GNAI3, PLCB4 | Autosomal dominant | “Question mark” ears, small mouth, prominent cheeks, anomalies of the temporomandibular joint and condyle |
| Cerebrocostomandibular syndrome | 117650 | Unknown | Autosomal dominant and recessive reported | Narrow thorax, rib anomalies, conductive hearing loss, growth restriction |
| Cerebrocostomandibular-like syndrome | 606973 | COG1 | Autosomal recessive | Costovertebral defects, microcephaly, growth restriction, developmental delay, brain anomalies, cryptorchidism |
| Talipes, ASD, PRS, and persistence of left superior vena cava (TARP) | 311900 | RBM10 | X-linked recessive | Talipes, congenital cardiac defects, persistence of left superior vena cava |
| Distal arthrogryposis-PRS (Illum syndrome) | 208155 | Unknown | Autosomal recessive | Joint contractures, autonomic instability, “whistling face,” nervous system classification |
| PRS, cleft mandible, limb anomalies (Richieri-Costa-Pereira syndrome) | 268305 | Unknown | Autosomal recessive | Short stature, hypoplastic thumbs, talipes, laryngeal malformations |
| Andersen-Tawil syndrome | 170390 | KCNJ2 | Autosomal dominant | Cardiac arrhythmias, periodic paralysis, low-set ears, short stature, digital anomalies |
| PRS + oligodactyly | 172880 | Unknown | Probably autosomal dominant | Oligodactyly |
| Fetal alcohol syndrome | — | N/A | Teratogenic | Microcephaly, growth and developmental delay, short palpebral fissures |
| Neuromuscular Conditions | ||||
| Congenital myotonic dystrophy | 160900 | DMPK | Autosomal dominant | Severe hypotonia, respiratory insufficiency |
| Carey-Fineman-Ziter syndrome | 254940 | Unknown | Probably autosomal recessive | Hypotonia, Moebius anomaly, growth delay, feeding difficulties, scoliosis |
| Chromosomal Abnormalities | ||||
| Trisomy 18 | — | Unknown | Sporadic chromosomal | Microcephaly, severe growth and cognitive impairment, prominent occiput, cardiac defects, low-set ears, short sternum, overlapping flexed fingers, prominent calcaneus |
| 22q11 microdeletion | 188400/192430 | TBX1 | Autosomal dominant chromosomal | Upslanting palpebral fissures, prominent tubular nose, conotruncal heart defects, renal and endocrine abnormalities, neuropsychiatric problems |
| 17q24 translocations and microdeletions | 261800 | SOX9 | Autosomal dominant chromosomal | Isolated PRS |
| 17q21.31q24.3 inversion and deletion | — | SOX9 | Autosomal dominant chromosomal | PRS with hypoploastic scapula and bilateral clubfeet |
| 2q33 microdeletion | 608148 | SATB2 | Autosomal dominant chromosomal | Growth restriction, feeding difficulties, downslating palpebral fissures, prominent nose, dental anomalies, behavioral problems, developmental delay |
| 4q terminal deletion | — | Unknown | Autosomal dominant chromosomal | Cardiac defects, mild developmental delay, but can be normal, stiff hypoplastic fifth fingers with hooked or volar nails |
| 1q duplication | — | Unknown | Autosomal dominant chromosomal | Bilateral fixed flexion deformities of fingers, cardiac and brain anomalies |
| Partial trisomy 11q | — | Unknown | Autosomal dominant chromosomal | Developmental delay, congenital heart defects |
| t(1;2)(p34;q33) | 613857 | FAF1 | Chromosomal | PRS in father and son with translocation disrupting FAF1 at 1p34 |
| 20p12.3 microdeletion | 112261 | BMP2 | Autosomal dominant chromosomal | Long philtrum, digital anomalies, hypotonia |
| 22q12 microdeletion | — | MN1 | Autosomal dominant chromosomal | Developmental delay, features of NF2 and Toriello-Carey due to contiguous gene deletion |
Stickler Syndrome.
Stickler syndrome is characterized by ocular findings (myopia, cataract, retinal detachment) and hearing loss (conductive and sensorineural) in addition to Pierre Robin sequence. Joint issues are also common (hypermobility, spondyloepiphyseal dysplasia, precocious osteoarthritis). Mutations in one of five genes are often present: COL2A1 , COL11A1 , and COL11A2 (autosomal dominant inheritance; COL2A1 most common, at 80-90%), as well as COL9A1 and COL9A2 (autosomal recessive; rare). Penetrance is complete but significant phenotypic variability occurs even within families. From 20% to 30% of patients with Pierre Robin sequence may have Stickler syndrome.
Cerebrocostomandibular Syndrome.
Cerebrocostomandibular syndrome (CCMS) is characterized by costovertebral anomalies (dorsal rib defects) in addition to Pierre Robin sequence. Rib defects often lead to a bell-shaped thorax and can result in flail chest. Other anomalies include growth retardation, scoliosis, dental anomalies, feeding issues, and conductive hearing loss. Severity is highly variable, although prognosis is often poor. This disorder is very rare, with slightly over 80 cases reported. Genetics are unknown, although possible defects in sonic hedgehog signaling have been implicated.



Treacher Collins Syndrome
Definition.
Treacher Collins syndrome is characterized by deficient generation of the lower two thirds of the face, including hypoplasia of the zygoma, maxilla, and mandible.
Synonyms.
Treacher Collins syndrome is also known as mandibulofacial dysostosis and Treacher Collins-Franceschetti syndrome.
Etiology.
The syndrome is due to abnormal development of the first and second branchial arches.
Incidence.
Incidence is 1 in 50,000 live births.
Diagnosis.
Affected individuals have maxillary and mandibular hypoplasia, choanal atresia/stenosis, downward slanting palpebral fissures, notching of the lower eyelid, sparse or absent eyelashes, microtia/absent external ear, middle ear hypoplasia, cleft palate ± lip, and dental anomalies.
Genetics.
Three causative genes have been found: TCOF1 and POLR1D (autosomal dominant inheritance; most common) as well as POLR1C (autosomal recessive; only 1% of cases). Sixty percent of cases result from de novo mutations. Significant phenotypic variability is seen, even within families. Penetrance is high, but cases of nonpenetrance have been reported.
Pathogenesis.
Mutations in the preceding genes lead to abnormal production of ribosomal RNA important in the development of the first and second branchial arches, leading structural facial anomalies.
Recurrence Risk.
Recurrence risk is 50% for the autosomal dominant genes if a parental mutation is present, low if mutation is de novo, and 25% for the autosomal recessive form.
Differential Diagnosis.
Differential diagnosis includes the other mandibulofacial dysostoses, such as Toriello syndrome, Bauru syndrome, Hedera-Toriello-Petty syndrome, and Guion-Almeida syndrome. Treacher Collins syndrome also shares some features of Nager syndrome, Miller syndrome, Goldenhar syndrome, and craniofacial microsomia.
Prognosis.
Intelligence is normal. Middle ear hypoplasia causes conductive deafness (40-50% of individuals). Vision loss can also occur (37%). Speech issues are common.
Management.
Intervention may be needed for the airway and feeding issues.
Van der Woude Syndrome
Definition.
Van der Woude syndrome (VWS) is included in a spectrum of IRF6 -related disorders ranging from isolated cleft lip/palate and popliteal pterygium syndrome. VWS is characterized by lower lip pits, cleft lip and palate, cleft uvula, and hypodontia.
Synonym.
VWS has been classified as types 1 and 2.
Incidence.
Incidence is 1 in 40,000 to 1 in 100,000 births. VWS is the most common form of syndromic clefting, accounting for 2% of all cases of cleft lip and palate.
Pathogenesis.
IRF6 (interferon regulatory factor 6) is a transcription factor involved in orofacial development.
Diagnosis.
Prenatal ultrasound examination can detect a cleft lip ± cleft palate, but is likely to miss an isolated cleft palate and lip pits.
Genetics.
VWS type 1 is caused by mutations in the IRF6 gene; type 2 is caused by mutations in the GRHL3 gene (5% of all VWS cases). Inheritance is autosomal dominant. Penetrance is high but can be incomplete. De novo mutations have been reported.
Recurrence Risk.
If a parental mutation is present, recurrence risk is 50%.
Differential Diagnosis.
Bartsocas-Pappas syndrome (popliteal pterygium syndrome, lethal type), Kabuki syndrome, BOFS, and isolated cleft lip and palate are considered in the differential diagnosis.
Prognosis.
Multiple surgeries are often needed to treat lip pitting and can lead to scarring and decreased ability to open the mouth. Psychomotor development is generally normal.

X-linked Opitz G/BBB Syndrome
Definition.
X-linked Opitz G syndrome (XLOS) is characterized by facial anomalies (hypertelorism, prominent forehead, widow’s peak, broad nasal bridge, anteverted nares), laryngeotracheoesophageal defects, and genitourinary anomalies (hypospadias, cryptorchidism, bifid scrotum).
Incidence.
Incidence is 1 in 50,000 to 1 in 100,000 males.
Pathogenesis.
Mutations in MID1 lead to altered mTORC1 signaling, which is thought to affect the development of ventral midline anatomic structures.
Diagnosis.
Diagnosis is most often based on clinical findings with abnormalities in ventral midline structures, as described previously. Prenatally, those cases associated with congenital heart defects and cleft lip are more likely to be detected.
Genetics.
XLOS is due to mutations in MID1 . Inheritance is X-linked. Many individuals are the only ones known to be affected within their families. Penetrance is high; wide phenotypic variability is seen even within families.
Recurrence Risk.
If the mother is a carrier, there is a 50% chance of transmitting the disease-causing mutation in each pregnancy. Sons who inherit the mutation will be affected. Daughters will be carriers with isolated hypertelorism.
Associated Anomalies.
Other anomalies include cleft lip/palate (50%), congenital heart defects, imperforate or ectopic anus, and midline brain defects.
Differential Diagnosis.
FG syndrome, craniofrontonasal dysplasia, and Mowat-Wilson syndrome should be considered.
Prognosis.
Intellectual disability and developmental delay are seen in 50% of affected males. Prognosis depends on the severity of the associated anomalies.
Management.
A multidisciplinary team is needed to manage the multiple anomalies.
Musculoskeletal System
Skeletal Dysplasias
Overall, skeletal dysplasias are seen in 2.4 per 10,000 births, and of these, 0.95 to 1.5 per 10,000 live births are lethal skeletal dysplasias. As a group, there are more than 450 skeletal dysplasias and the specific disorder can be difficult to determine based on prenatal ultrasound findings alone. A genetic cause has been identified for more than 50% of skeletal dysplasias; many are due to de novo mutations. Those with autosomal recessive inheritance often occur without a family history.
In the prenatal setting, particular emphasis should be given to identifying those disorders with neonatal or infantile lethality. Specific details should be obtained on prenatal ultrasound examination if a skeletal dysplasia is suspected, including length and appearance of long bones, facial features, spine, and extremity anomalies, as well as femur/foot ratio. Measurements that can assist in predicting neonatal lethality include chest circumference/abdominal circumference ratio and femur length/abdominal circumference ratio.
Of the lethal skeletal dysplasias, the most common are thanatophoric dysplasia (TD) and osteogenesis imperfecta (type 2), followed by achondrogenesis. Together, these three disorders represent 40% to 60% of all lethal skeletal dysplasias. Other relatively common lethal skeletal dysplasias include campomelic dysplasia, hypophosphatasia congenita, and short-rib polydactyly syndromes. Lethal disorders seen more rarely include atelosteogenesis, rhizomelic chondrodysplasia punctata, and fibrochondrogenesis. The most common nonlethal skeletal dysplasia is achondroplasia. These disorders are each reviewed in more detail in this section.
Thanatophoric Dysplasia
Definition.
TD is the most common lethal skeletal dysplasia and is characterized by severe rhizomelia, small thorax, normal trunk length, normal bone mineralization, no fractures, thickened redundant skin, and platyspondyly (flattened vertebral bodies). TD is divided into two subtypes:
- •
Type 1: TD1 is the more common form. The femurs have a “telephone receiver” shape, and frontal bossing and midface hypoplasia are present. Cloverleaf skull is not seen. The hands and feet are normal, but the fingers are short.
- •
Type II: TD2 is the less common form. Femurs are straight with flared metaphyses, and a cloverleaf skull is present (due to craniosynostosis of the lambdoid and coronal sutures).
Incidence.
Incidence is 1 in 20,000 births.
Etiology.
Mutations in FGFR3 are gain-of-function mutations that lead to a constitutively active protein. This leads to chondrocyte proliferation and premature maturation of bone.
Genetics.
TD1 is caused most frequently by mutations in the fibroblast growth factor receptor 3 gene ( FGFR3 ); the most common mutations are R248C and Y373C. TD2 is also caused by mutations in FGFR3 , specifically the mutation K650E . Inheritance is autosomal dominant. Because the disorder is lethal, new diagnoses are generally the result of de novo mutations. Advanced paternal age has been associated with an increased risk.
Recurrence Risk.
Recurrence risk is low because most mutations are de novo; germline mosaicism remains a possibility but has not been reported in individuals without signs of skeletal dysplasia.
Diagnosis.
Prenatal findings include severe rhizomelia (seen as early as 12-14 weeks) and a hypoplastic thorax. Craniofacial dysmorphisms include macrocephaly, frontal bossing, midface hypoplasia, and cloverleaf skull. Other features include brachydactyly, platyspondyly, hypotonia, and redundant skinfolds. Other brain anomalies may be seen including polymicrogyria, deep and transverse temporal sulci, and temporal lobe enlargement. Polyhydramnios is seen in approximately 50% of cases. Increased nuchal translucency may be present.
Differential Diagnosis.
The differential diagnosis includes other types of short-limbed dwarfism and disorders with a cloverleaf skull (such as Apert, Crouzon, Pfeiffer, and Carpenter syndromes). The features can closely resemble homozygous achondroplasia with autosomal recessive inheritance; it can be distinguished from TD by the presence of positive carrier status for heterozygous FGFR3 mutations in each parent in the case of achondroplasia.
Prognosis.
In general, affected infants die shortly after birth of respiratory insufficiency due to a small thorax and lung hypoplasia.
Management.
The option of pregnancy termination should be offered before viability. Sonographic evaluation of hydrocephalus is recommended because it can lead to malpresentation and difficult delivery. If massive hydrocephalus has developed, cephalocentesis or elective cesarean delivery should be considered to avoid maternal trauma.


Osteogenesis Imperfecta
Definition.
Osteogenesis imperfecta (OI) is a heterogeneous group of genetic disorders (classically divided in four types: I, II [congenital], III, and IV) characterized by severe bone fragility, leading to abnormal ossification and multiple fractures. The clinical features range from perinatal lethality (type II) to severe skeletal deformities to those with minimal features. The four types are described as follows:
- •
Type I: classic nondeforming OI with blue sclerae. Deformities are not seen prenatally.
- •
Type II: perinatal lethal OI. This is the most severe form and can be seen prenatally.
- •
Type III: progressively deforming OI. Some features can be seen prenatally.
- •
Type IV: common variable OI with normal sclerae. Deformities are not generally seen prenatally.
Synonyms.
OI is also called brittle bone disease.
Incidence.
The incidence is 6 to 7 per 100,000 for all types of OI, with types I and IV representing more than one half of all cases. Types II and II each have an incidence of 1 to 2 per 100,000.
Etiology.
COL1A1 and COL1A2 encode for type I collagen (found in skin, ligaments, tendons, demineralized bone, and dentine). Defective production of type I collagen leads to decreased bone mineralization and bone fragility.
Genetics.
OI is due to mutations in COL1A1 or COL1A2 . Inheritance is autosomal dominant. Approximately 60% of cases of type I and IV OI are caused by de novo mutations, whereas almost 100% of cases of type II and III OI are caused by de novo mutations. Penetrance is complete, but expression can vary significantly.
Recurrence Risk.
Recurrence risk depends on whether the mutation was inherited or de novo. Gonadal mosaicism may be present in 3% to 5% of cases with apparent de novo mutations. Less commonly, OI can also be autosomal recessive.
Diagnosis.
Type II is the most severe form and affected individuals usually die in utero or shortly after birth from severe fractures and pulmonary hypoplasia. Key prenatal findings include severe micromelia (femur length more than 3 standard deviations [SD] below the mean), small thorax, normal head circumference, short trunk, decreased bone mineralization (including decreased ossification of the skull), and multiple bone fractures. Multiple fractures may lead the long bones to appear angulated. Both cortices of a bone are often seen. Some findings can be seen as early as 13 to 15 weeks’ gestation.
The other types of OI may not be detected prenatally. Type I is characterized by blue sclerae and normal stature. The first fractures generally occur in infancy. Progressive hearing loss is present in 50% of adults. Type III is apparent at birth; fractures may be caused just by handling the infant. Rib fractures can lead to pulmonary failure in the first few weeks or months of life. Those who survive generally need assistance for mobility and are extremely short. Hearing loss often begins in the teenage years. Type IV is characterized by mildly short stature, adult-onset hearing loss, and normal-to-gray sclerae; phenotype is the most variable. Dentinogenesis imperfecta is common.
Differential Diagnosis.
The differential diagnosis includes hypophosphatasia (infantile form), achondrogenesis, and other short-limbed dwarfisms.
Prognosis.
Type II OI is uniformly lethal. The other forms develop after birth and are characterized by progressive deformities of the long bones and short stature.
Management.
Owing to the uniformly fatal outcome, termination of pregnancy should be offered when type II is diagnosed early in pregnancy and palliative care provided if delivery is undertaken. If continuation of the pregnancy is chosen, cesarean delivery is often recommended for nonlethal OI for prevention of fetal and maternal trauma.

Related posts:
 Ultrasound Evaluation of the Fetal Central Nervous System
Ultrasound Evaluation of the Fetal Central Nervous System
 Obstetric Ultrasound Imaging and the Obese Patient
Obstetric Ultrasound Imaging and the Obese Patient
 Evaluation of Hydrops Fetalis
Evaluation of Hydrops Fetalis
 Fetal Cardiac Measurements and Doppler Assessment
Fetal Cardiac Measurements and Doppler Assessment
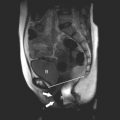 Ultrasound and Magnetic Resonance Imaging in Urogynecology
Ultrasound and Magnetic Resonance Imaging in Urogynecology
 Gynecologic Sonography in the Pediatric and Adolescent Patient
Gynecologic Sonography in the Pediatric and Adolescent Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


