Urinary Tract
Imaging plays an important role in the evaluation of suspected or known abnormalities of the urinary tract in infants and children. Excretory urography, once the cornerstone for imaging renal abnormalities, is now rarely used in this evaluation since the advent of newer imaging modalities. Sonography, computed tomography (CT), magnetic resonance imaging (MRI), and nuclear scintigraphy have become the major imaging studies for evaluating the upper urinary tract in children (1,2,3,4). Angiographic and interventional techniques have a more limited role in renal imaging, but on occasion they aid in diagnosis and treatment.
The common clinical problems that initiate imaging examinations are abnormal prenatal sonography (usually hydronephrosis, multicystic dysplastic kidney, and congenital anomalies), palpable abdominal masses, and urinary tract infection. These abnormalities are detailed in this chapter. Other abnormalities discussed herein include medical and vascular renal diseases, stone diseases, and trauma.
NORMAL ANATOMY
At sonography, the neonatal and infant kidneys have three unique features that differentiate them from the kidneys of older children and adults (1). First, the echogenicity of the renal cortex is increased and usually is slightly greater that of the liver or spleen (Fig. 8.1). The more premature the infant, the higher the likelihood is that the renal cortex will be hyperechoic. The second difference is that in the infant, the medullary pyramids are moderately or markedly hypoechoic and strikingly prominent (Fig. 8.1). The third unique feature of the kidney in neonates and young infants is that the renal sinus is not as echogenic as in older adolescents and adults, because of the paucity of fat in this area (Fig. 8.1).
Neonatal and infant kidneys have three unique features that differentiate them from kidneys of older children.
By the end of the first year of life, the renal cortex usually becomes hypoechoic to the adjacent liver or spleen. The normal medullary pyramids are minimally hypoechoic. The renal sinus appears as a central echogenic area (Fig. 8.2).
The resistive index (RI) varies with patient age (1). In neonates, the RI can be as high as 0.85 (range, 0.56 to 0.85). The RI decreases in the postnatal period, and by the middle of the first decade of life, it is equal to or less than 0.70.
The resistive index (RI) varies with patient age.
ANATOMIC VARIATIONS
FETAL LOBULATION
The normal kidney results from fusion of embryonic renunculi or lobes, which develop during the second trimester. Each renunculus consists of a large central pyramid that
contains renal tubules and that is enveloped by a layer of cortex containing glomeruli. Fusion of the renunculi produces a single kidney with lobulated or irregular margins, referred to as fetal lobulation (5). During later in utero development, the renal surface becomes smoother, although occasionally fetal lobulation persists in postnatal life (Fig. 8.3). Fetal lobulation should not be mistaken for renal scarring, which is associated with parenchymal thinning.
contains renal tubules and that is enveloped by a layer of cortex containing glomeruli. Fusion of the renunculi produces a single kidney with lobulated or irregular margins, referred to as fetal lobulation (5). During later in utero development, the renal surface becomes smoother, although occasionally fetal lobulation persists in postnatal life (Fig. 8.3). Fetal lobulation should not be mistaken for renal scarring, which is associated with parenchymal thinning.
 Figure 8.1 Normal renal anatomy. Neonatal kidney, longitudinal scans. Note that the echogenicity of the cortex equals that of the adjacent liver (L). The pyramids (arrowheads) are prominent. The central renal sinus (arrow) is barely discernable. |
Fetal lobulation should not be mistaken for renal scarring.
COLUMN OF BERTIN
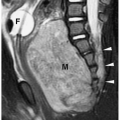
The adjacent cortices of the contiguous renunculi fuse to form a thick layer of cortex, termed a column of Bertin (Fig. 8.4). This column occasionally hypertrophies and causes splaying of the calyces. The column of Bertin has an echogenicity, attenuation value, and signal intensity that is equal to or slightly greater than that of the cortex. It extends from the renal cortex to the renal sinus and is located between two medullary pyramids. The absence of renal contour abnormality is a useful feature in differentiating a hypertrophied column of Bertin from a true mass.
Column of Bertin is located between two medullary pyramids.
RENAL DUPLICATION
Collecting system duplication is the most common renal anomaly (5) and may be partial or complete. The spectrum of partial duplication ranges from a bifid renal pelvis to
Y-shaped ureters with the two ureters fusing somewhere along their course. A single common distal ureter enters the bladder. With complete ureteral duplication, the kidney has two pelvicalyceal systems and two ureters with separate insertions. Complete duplications are discussed in more detail later in the chapter. Partial duplication anomalies are generally not clinically significant and are found incidentally on sonography or CT. The nonobstructed duplex kidney is larger than the normal single system kidney and it has two separate renal sinus echo complexes on sonography and two renal pelvices on CT (Fig. 8.5).
Y-shaped ureters with the two ureters fusing somewhere along their course. A single common distal ureter enters the bladder. With complete ureteral duplication, the kidney has two pelvicalyceal systems and two ureters with separate insertions. Complete duplications are discussed in more detail later in the chapter. Partial duplication anomalies are generally not clinically significant and are found incidentally on sonography or CT. The nonobstructed duplex kidney is larger than the normal single system kidney and it has two separate renal sinus echo complexes on sonography and two renal pelvices on CT (Fig. 8.5).
Collecting system duplication is the most common renal anomaly.
 Figure 8.2 Normal renal anatomy, 3-year-old child. The renal cortex is less echogenic than the liver (L). The renal pyramids are less prominent, and the central renal sinus (arrow) is moderately echogenic. |
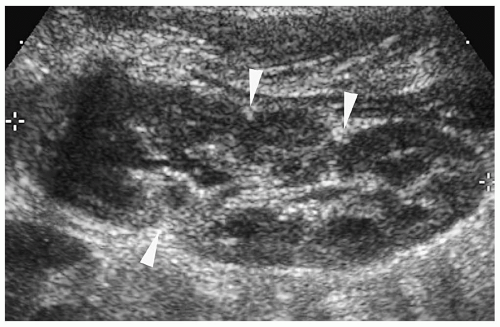 Figure 8.3 Anatomic variant, fetal lobulations. Longitudinal scan of the right kidney shows lobulated renal margin (arrowheads). Fetal lobulations lie between the renal pyramids or calyces, unlike cortical scars that lie directly over the calyces. |
The duplex kidney is larger than the single system kidney.
CONGENITAL ANOMALIES
MALPOSITIONED KIDNEYS
Simple Ectopia
In simple renal ectopia, the kidney and ureter are on their expected sides of the spine. The ectopic kidney is most commonly located in the pelvis. It is usually small and malrotated and frequently has a dysmorphic configuration, such as a pancake, disk, or lump shape (Fig. 8.6).
The ectopic kidney is most commonly located in the pelvis.
Crossed Fused Ectopia
In crossed fused ectopia, both kidneys are located on the same side of the spine. The crossed ectopic kidney is smaller than the normal kidney and malrotated, and it usually lies caudal to the orthotopic kidney. Typically, the upper pole of the ectopic kidney is fused to the lower pole of the orthotopic unit (Fig. 8.7). Both distal ureters are normally located. The ureter
draining the ectopic kidney returns enters the bladder at the contralateral trigone, whereas the ureter of the orthotopic component enters the ipsilateral trigone. There is an increased incidence of multicystic dysplasia, hydronephrosis, infection, and calculus formation in the crossed unit.
draining the ectopic kidney returns enters the bladder at the contralateral trigone, whereas the ureter of the orthotopic component enters the ipsilateral trigone. There is an increased incidence of multicystic dysplasia, hydronephrosis, infection, and calculus formation in the crossed unit.
The crossed ectopic kidney is smaller than the normal kidney and malrotated, and usually lies caudal to the orthotopic kidney.
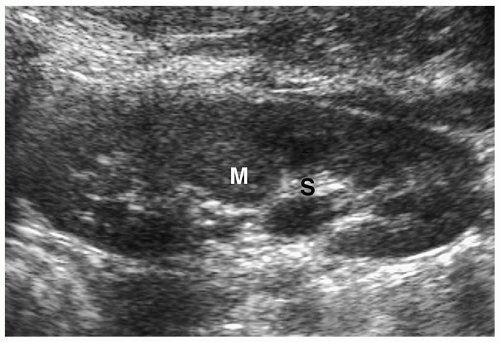 Figure 8.4 Anatomic variant, hypertrophied column of bertin. Longitudinal sonogram shows a mass-like structure (M) extending from the renal cortex to the renal sinus (S). The hypertrophied column typically has an echogenicity similar to the remainder of the kidney and often displaces the sinus fat and pyramids. |
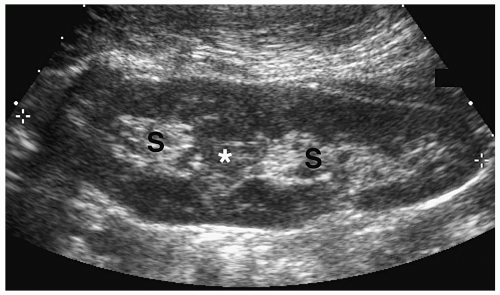 Figure 8.5 Anatomic variant, renal duplication. Longitudinal sonogram shows an elongated kidney (cursors) with a prominent central cleft of normal appearing parenchyma (*) separating two echodense renal sinuses (S). |
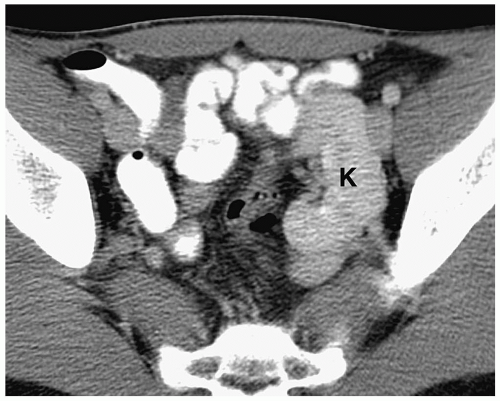 Figure 8.6 Ectopic kidney. Contrast-enhanced CT scan in the same patient as in Figure 8.5 shows an ectopically positioned pelvic kidney (K). |
 Figure 8.7 Crossed fused ectopia. A. Oblique scan of the right lower flank in a neonate demonstrates fusion of the upper pole of the left kidney (LK) with the lower pole of the right kidney (RK). (P, renal pelvis) (S, spine). B. Contrast-enhanced CT in a teenager shows fusion of the left kidney (LK) with the lower pole of the right kidney (RK). |
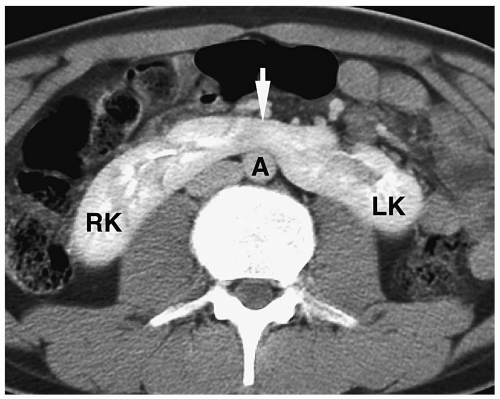 Figure 8.8 Horseshoe kidney. Contrast-enhanced CT shows an isthmus of tissue (arrow) anterior to the aorta (A) connecting the lower poles of the kidneys (RK, right kidney; LK, left kidney). |
Horseshoe Kidney
Horseshoe kidney is the most common renal fusion anomaly. Typically, the lower poles of the kidneys are fused and connected by an isthmus of tissue. Associated anomalies are common and include uteropelvic junction obstruction, vesicoureteral reflux, duplicated collecting systems, and renal dysplasia. There is an increased risk of infection and stone formation. There also is a slightly increased incidence of Wilms tumor and renal cell carcinoma. Imaging findings include anteriorly located renal pelves, medial orientation of the lower poles of the kidneys, and an isthmus of tissue crossing the midline anterior to the great vessels (Fig. 8.8).
Horseshoe kidney is the most common renal fusion anomaly.
RENAL AGENESIS
Renal agenesis is diagnosed when there is absence of a kidney, compensatory hypertrophy of the contralateral kidney, and adjacent organ displacement. On the right, the proximal small bowel, hepatic flexure, liver, or pancreatic head fill the empty renal fossa. On the left, small bowel loops, the splenic flexure, or the pancreatic tail fill the corresponding space. Other findings of renal agenesis are thin elongated adrenal glands and absence of the ipsilateral renal artery and vein (see Fig. 9.2, Chapter 9). The contralateral kidney is normal in size at birth, but within 6 to 12 months of life begins to show compensatory hypertrophy. Associated genital anomalies are common, including absence of the seminal vesicles or vas deferens, seminal vesicle cysts, undescended testes, uterine and/or vaginal duplication, and imperforate vagina with secondary hydrometrocolpos.
Bilateral renal agenesis is incompatible with life, and affected infants are either stillborn or die in the immediate postnatal period. Almost all patients have pulmonary hypoplasia as a result of oligohydramnios and uterine compression of the fetal thorax. The kidneys and renal arteries are absent, and the bladder is small or absent.
CONGENITAL HYDRONEPHROSIS AND HYDROURETER
Congenital hydronephrosis may be secondary to mechanical or functional causes (6,7). Mechanical causes of obstruction include ureteropelvic junction obstruction, ureterovesical
junction obstruction, duplex collecting system anomalies, and posterior urethral valves. Functional causes include prune belly syndrome and vesicoureteral reflux. Congenital hydronephrosis is nearly always diagnosed on prenatal sonography. Occasionally, patients present with a palpable abdominal mass or with complications of obstruction, including flank pain, hematuria, and urinary tract infection. In these cases, sonography is usually the initial imaging study, although on occasion CT may be the first study performed.
junction obstruction, duplex collecting system anomalies, and posterior urethral valves. Functional causes include prune belly syndrome and vesicoureteral reflux. Congenital hydronephrosis is nearly always diagnosed on prenatal sonography. Occasionally, patients present with a palpable abdominal mass or with complications of obstruction, including flank pain, hematuria, and urinary tract infection. In these cases, sonography is usually the initial imaging study, although on occasion CT may be the first study performed.
GENERAL IMAGING APPROACH
When intrauterine hydronephrosis is diagnosed, a postpartum sonogram is obtained 4 to 5 days after delivery (8). A sonogram performed earlier may be falsely negative or may underestimate the severity of hydronephrosis. If the postnatal sonogram shows moderate or severe hydronephrosis, further evaluation is usually undertaken and a voiding cystourethrogram (VCUG) and diuretic scintigraphy are performed. Voiding cystography assesses the presence of reflux and diuretic scintigraphy, usually with 99mTc-metacaptoacetyltriglycine (MAG3), assesses differential renal function. When the initial postnatal sonogram is normal or shows mild dilatation, it is generally repeated at 6 weeks of age. If follow-up sonography shows mild hydronephrosis, a VCUG is performed.
The following discussion emphasizes the sonographic findings of congenital hydronephrosis. Although congenital hydronephrosis can be seen on CT and MRI, these studies are not usually necessary to evaluate congenital urinary tract obstruction.
URETEROPELVIC JUNCTION OBSTRUCTION
Ureteropelvic junction (UPJ) obstruction is the most common cause of congenital urinary tract obstruction. It is believed to be due to abnormal development of a short segment of the ureteral smooth muscle at the ureteropelvic junction. Extrinsic factors, such as bands, adhesions, or aberrant vessels crossing the upper ureter or renal pelvis, have also been implicated as causes of obstruction. Classic sonographic findings are a dilated upper collecting system and a normal ureter and bladder (Fig. 8.9). Characteristically, the renal pelvis dilates more than the calyces. If the hydronephrosis is severe, there may be parenchymal thinning.
Sonographic findings in UPJ are a dilated upper collecting system and normal ureter and bladder.
UPJ obstruction may coexist with other urinary tract abnormalities, including vesicoureteral reflux, ureterovesical junction obstruction, horseshoe kidney, and duplex kidney. In duplex kidney, the obstruction usually occurs in the upper pole moiety. Because of the coexistence of reflux, a VCUG should be performed if the sonogram suggests UPJ obstruction.
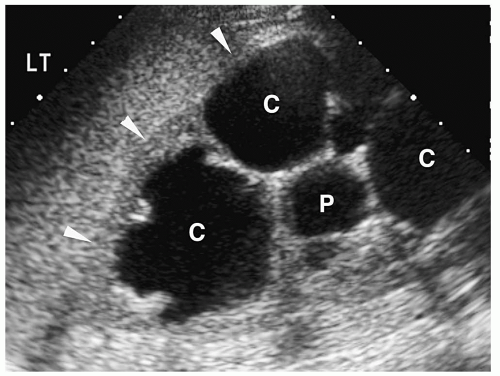 Figure 8.9 Ureteropelvic junction obstruction. A. Longitudinal sonogram of the left kidney demonstrates marked dilatation of the calyces (C) and renal pelvis (P). A thin rim of parenchyma (arrowheads) surrounds the dilated collecting system. No ureter was identified on transverse scan through the urinary bladder. |
 Figure 8.10 Megaureter. A. Longitudinal sonogram of the left kidney (cursors) in a 3-month-old girl reveals mildly dilated calyces (arrowheads) and renal pelvis (P). B. Longitudinal image through the lower pelvis shows a markedly dilated distal left ureter (U), tapering prior to entering the bladder (B). C. Image from a nuclear medicine lasix renogram shows persistent left hydroureteronephrosis (LK, left kidney; U, ureter; B, bladder) after the radiopharmaceutical has cleared to right kidney (RK). |
The treatment of choice in infants and young children is a dismembered pyeloplasty where the redundant renal pelvis is trimmed and reinserted into the ureter. In some adolescent patients, endopyelotomy has replaced pyeloplasty for repair of ureteropelvic junction obstruction.
PRIMARY MEGAURETER
Primary megaureter refers to ureteral dilatation above a short aperistaltic juxtavesical segment of ureter that has a normal insertion into the trigone (9). The cause of aperistalsis is unclear, but it may be due to an absence of ganglion cells in the distal ureter. Sonographic findings include ureterectasis and variable degrees of pelviectasis and caliectasis (Fig. 8.10). The dilatation of the proximal ureter is often less than that of the distal ureter. Patients with moderate and severe primary megaureter are usually treated with ureteral tapering and reimplantation. Patients with mild megaureter are followed clinically and seldom undergo reimplantation.
Findings in megaureter include ureterectasis and variable degrees of pelviectasis and caliectasis.
URETERAL DUPLICATION
As noted above, ureteral duplication can be partial or complete. The latter is more likely to be obstructed. In complete ureteropelvic duplication, each renal moiety has its
own ureteral orifice in the bladder. The ureter of the upper pole moiety inserts medial and inferior to the ureter draining the lower pole moiety (Weigert-Meyer rule). Complete duplication anomalies are more common in girls than in boys. If the diagnosis is not made in utero, patients often present later in life with signs of urinary tract infection. Girls also present with incontinence if the ureter inserts below the external sphincter.
own ureteral orifice in the bladder. The ureter of the upper pole moiety inserts medial and inferior to the ureter draining the lower pole moiety (Weigert-Meyer rule). Complete duplication anomalies are more common in girls than in boys. If the diagnosis is not made in utero, patients often present later in life with signs of urinary tract infection. Girls also present with incontinence if the ureter inserts below the external sphincter.
In renal duplication, the upper pole ureter inserts medial and inferior to the lower pole ureter (Weigert-Meyer rule).
The sonographic features of an obstructed ureteral duplication include a hydronephrotic upper pole moiety, a normal or mildly dilated lower pole moiety, parenchymal loss in the upper pole, and two separate ureters (Figs. 8.11 and 8.12). Rarely, the upper pole is dysplastic. A dysplastic upper pole appears highly echogenic with small cysts.
The ureter from the upper pole renal moiety may insert into the vestibule, vagina, or urethra in girls and into the posterior urethra, epididymis, or the seminal vesicles in boys. In both sexes, the ectopic ureters also may drain into the bladder inferomedial to the normal location. When it inserts into the bladder, the distal part of the ectopic ureter has a cyst-like appearance, termed an ectopic ureterocele. The ureter from the lower pole moiety is orthotopic and inserts into the trigone. It usually has a perpendicular course rather than the normal oblique course, predisposing it to reflux.
 Figure 8.11 Ureteral duplication with an ectopic ureter. A. Longitudinal sonogram of the left kidney in a newborn boy with prenatal “hydronephrosis” reveals a dilated upper (UP) pole collecting system. There is mild dilatation of the renal pelvis in the lower pole collecting system. B. Longitudinal sonogram through the bony pelvis shows a dilated ureter (U) extending below the bladder (B) base. No ureterocele is identified in the bladder. C. Voiding cystourethrogram (VCUG) image shows reflux into the left ureter (U), which inserted ectopically (arrow) into the posterior urethra below the bladder neck. |
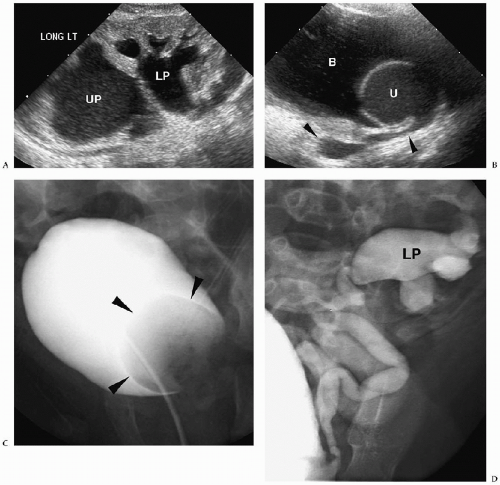 Figure 8.12 Ureteral duplication with an ectopic ureterocele. A. Longitudinal sonogram of the left kidney in a newborn with prenatal hydronephrosis demonstrates a duplex kidney with hydronephrosis of both the upper (UP) and lower (LP) poles. The upper pole collecting system has echogenic debris. B. Longitudinal sonogram through the bladder (B) shows a dilated left ureter (arrowheads) emptying into an ectopic ureterocele (U) filled with echogenic debris. C. Filling image from the voiding cystourethrogram (VCUG) shows a large left ureterocele (arrowheads). D. Later VCUG image shows grade IV vesicoureteral reflux into the lower pole (LP) of the left kidney (“drooping lily” sign). |
On sonography and voiding cystourethrography, the ureterocele appears as a round, thin-walled, urine- or contrast-filled intravesical mass (see Fig. 8.12). Occasionally, the ureterocele will evert or intussuscept into or adjacent to its own ureter, mimicking a bladder diverticulum (Fig. 8.13).
Ectopic ureteroceles are treated by endoscopic incision and unroofing. Heminephrectomy may be performed in patients with severe upper pole hydronephrosis and poor renal
function. In patients with mild hydronephrosis, the ureter from the upper pole moiety may be reimplanted into the ureter from the lower pole moiety.
function. In patients with mild hydronephrosis, the ureter from the upper pole moiety may be reimplanted into the ureter from the lower pole moiety.
 Figure 8.13 Everting ureterocele. A. Voiding cystourethrogram in a child with right upper pole hydronephrosis shows a filling defect (arrowheads) within the bladder representing a ureterocele. B. Later image after voiding shows that the ureterocele has everted (arrowheads) outside the bladder (B) and vesicoureteral reflux into the right kidney (RK). |
POSTERIOR URETHRAL VALVES
Posterior urethral valves are the most common cause of urethral obstruction in newborn boys. Pathologically, urethral valves are folds of embryonic tissue. They may or may not have a small aperture. Most insert at the level of the membranous urethra, but they may insert above or below the verumontanum.
Posterior urethral valves are the most common cause of urethral obstruction in newborn boys.
Characteristic sonographic findings include bilateral hydronephrosis and hydroureter, parenchymal thinning, a thickened bladder wall, and posterior urethral dilatation (Fig. 8.14). The valve appears as an area of linear echogenicity within the dilated posterior urethra.
Sonographic findings include hydronephrosis and hydroureter, thickened bladder wall, and posterior urethral dilatation.
Other findings include urinary ascites, subcapsular or perirenal urinomas, and renal dysplasia. Urinary ascites and urinoma formation result when there is calyceal rupture. The perforation serves as a method to decompress the collecting system; thus, hydronephrosis may be minimal or barely apparent. Renal dysplasia is the result of high-pressure in utero reflux. Dysplastic kidneys are small and echogenic and they may contain cortical cysts (Fig. 8.15).
Cystography remains the standard for confirming the diagnosis. Cystographic findings include a thickened and trabeculated bladder, a dilated posterior urethra, a narrow bladder neck, vesicoureteral reflux, and reflux of contrast into prostatic ducts or the prostatic utricle. The valve may be seen as a linear filling defect in the posterior urethra (see Fig. 8.14). Treatment is early cystotomy followed later by valve resection.
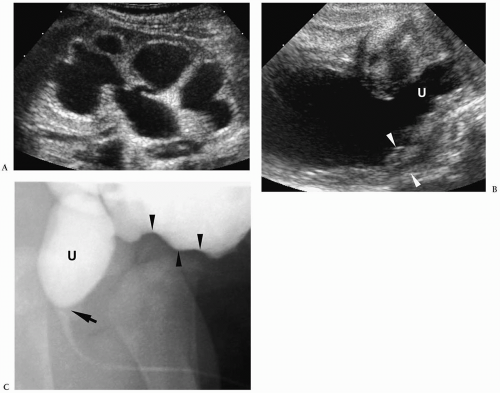 Figure 8.14 Posterior urethral valves. A. Longitudinal sonogram of the right kidney in a newborn boy with prenatal hydronephrosis reveals marked pelvicaliceal dilatation. B. Longitudinal image through the lower pelvis demonstrates bladder wall thickening (between arrowheads). The wall thickness measured 7 mm (normal thickness = 3 mm). The posterior urethra (U) is dilated. C. Voiding image from the VCUG shows the posterior urethra (U) dilated down to a thin-filling defect (arrow) representing the posterior urethral valve. The bladder wall is irregular (arrowheads) indicating trabeculation. |
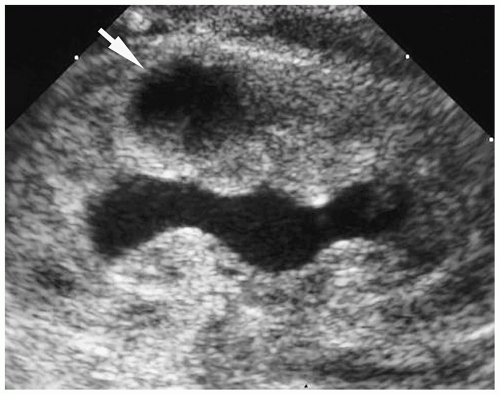 Figure 8.15 Posterior urethral valves, renal dysplasia. Longitudinal image of the left kidney in a newborn shows an echogenic kidney with mild hydronephrosis, loss of normal cortical medullary differentiation, and a cortical cyst (arrow), indicating renal dysplasia. |
PRUNE-BELLY SYNDROME
The prune belly, or Eagle-Barret syndrome is characterized by the triad of hypoplastic or absent abdominal wall musculature, urinary tract anomalies, and cryptorchidism. It occurs almost exclusively in male infants.
The prune belly, syndrome is characterized by the triad of hypoplastic or absent abdominal wall musculature, urinary tract anomalies, and cryptorchidism.
There are two basic groups of patients with prune-belly syndrome. The first group has urethral valves or atresia and bilateral renal dysplasia, which results in death shortly after birth. The second group has varying degrees of hydronephrosis, usually moderate to severe. The bladder is large, and a urachal diverticulum is often present. Infants in group 2 have a functional abnormality of bladder emptying but no urethral obstruction. They survive the neonatal period, but they have recurrent urinary tract infections or failure to thrive.
Plain radiographs show bulging flanks, flaring of the lower rib ends, and gaseous distention of bowel. Sonographic findings include varying degrees of hydronephrosis, dilated tortuous ureters, a large urinary bladder, a urachal diverticulum, a dilated posterior and/or anterior urethra (megalourethra), and a dilated prostatic utricle (Fig. 8.16). The
renal parenchyma may be echogenic and contain small cysts due to associated dysplasia. Cystographic findings include a large bladder, urachal diverticulum, wide open bladder neck, megalourethra, and vesicoureteral reflux (Fig. 8.17).
renal parenchyma may be echogenic and contain small cysts due to associated dysplasia. Cystographic findings include a large bladder, urachal diverticulum, wide open bladder neck, megalourethra, and vesicoureteral reflux (Fig. 8.17).
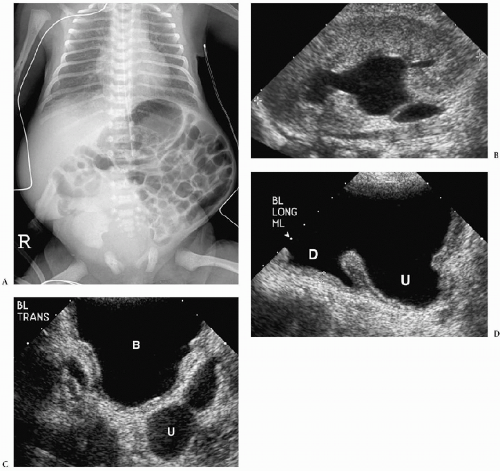 Figure 8.16 Prune-belly syndrome. A. Radiograph of a newborn shows bulging flanks secondary to the absence of abdominal wall musculature. B. Longitudinal sonogram of the left kidney (cursors) reveals a dilated collecting system. The right kidney had a similar appearance. C. Transverse sonogram shows a dilated ureter (U) posterior to the bladder (B). D. Longitudinal sonogram through the bladder shows a dilated posterior urethra (U) and a bladder diverticulum (D). |
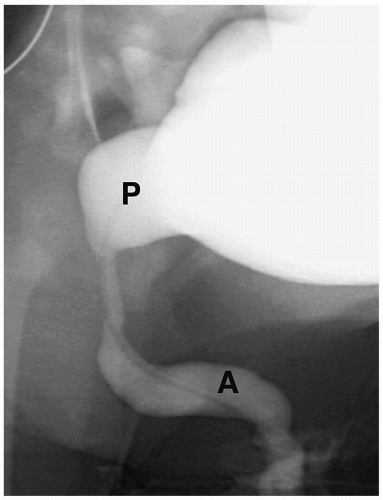 Figure 8.17 Prune-belly syndrome. Image during voiding shows a large, smooth-walled bladder and megalourethra with dilated anterior (A) and posterior (P) segments. |
OTHER CAUSES OF HYDRONEPHROSIS
Ectopic insertion of a ureter from a single collecting system is more common in boys than in girls. Imaging studies show ectopic termination of a solitary ureter. The sites of insertion are similar to those described for ureteropelvic duplications.
RENAL CYSTIC DISEASE
Cystic diseases of the kidney can be inherited or sporadic, unilateral or bilateral, and symptomatic at birth or detected later in life (10,11). This section will approach the differential diagnosis from the standpoint of unilateral and bilateral disease.
UNILATERAL CYSTIC DISEASE
Simple Cysts
Simple cysts are rare in children, with an incidence of less than 1%. Pathologically, they are unilocular masses lined by a single layer of flattened epithelium and contain clear serous fluid. Simple cysts arise in the renal cortex, do not communicate with the collecting system, and are more often solitary than multiple. They are usually asymptomatic and
detected during imaging examinations performed for other indications. However, very large cysts can present as palpable abdominal masses. Sonographic, CT, and MR features include a smooth, round shape; imperceptible walls; and homogenous fluid contents. The echogenicity, attenuation value, and signal intensity are equal to that of water. The cysts are avascular and do not enhance after administration of intravenous iodinated contrast medium or gadolinium (Fig. 8.18).
detected during imaging examinations performed for other indications. However, very large cysts can present as palpable abdominal masses. Sonographic, CT, and MR features include a smooth, round shape; imperceptible walls; and homogenous fluid contents. The echogenicity, attenuation value, and signal intensity are equal to that of water. The cysts are avascular and do not enhance after administration of intravenous iodinated contrast medium or gadolinium (Fig. 8.18).
Simple renal cysts are rare in children.
 Figure 8.18 Simple renal cyst. Contrast-enhanced CT scan shows a water-attenuation mass (M) with smooth, well-defined walls and no soft tissue component. |
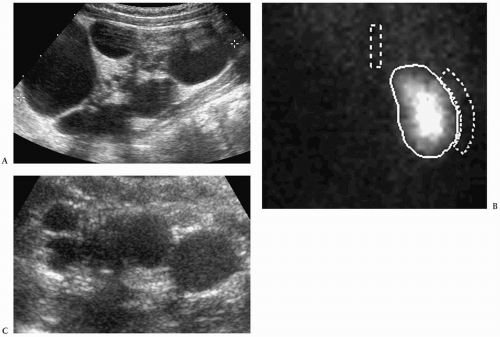 Figure 8.19 Multicystic dysplastic kidney. A. Longitudinal sonogram in a newborn girl reveals multiple oval and round cysts of varying sizes in the right renal fossa. There is no central renal pelvis or normal renal parenchyma noted. B. Scintigraphy shows unilateral left renal uptake with absence of radiopharmaceutical uptake in the dysplastic right kidney. C. Longitudinal sonogram at 6 months shows interval decrease in number and size of the cysts. |
Calyceal diverticula need to be distinguished from simple cysts. The former are cystic spaces that are lined by transitional epithelium and communicate with the collecting system through a narrow orifice. Most diverticula demonstrate some filling with contrast-enhanced urine, usually best seen on delayed CT scans.
Multicystic Dysplastic Kidney
Multicystic dysplastic kidney is a nonhereditary developmental anomaly. Histologically, there are noncommunicating cysts of various sizes, separated by tissue containing primitive dysplastic elements. Contralateral renal abnormalities are common and include vesicoureteral reflux, ureteropelvic junction obstruction, primary megaureter, and ureteropelvic disruption. Most cases are diagnosed on prenatal sonography.
The sonographic features of the classic multicystic dysplastic kidney are multiple cysts of varying size with a random distribution, absence of communication between the cysts, no discernible renal pelvis or sinus, and absent or dysplastic renal parenchyma (Fig. 8.19). Calcifications may be noted in the cyst walls or in the septations between cysts. The contralateral kidney usually shows compensatory hypertrophy. Rarely, multicystic dysplastic kidney may appear as a solitary cystic mass in the renal fossa.
Sonographic features of multidysplastic kidney are multiple cysts, absence of communication between the cysts, no discernible renal pelvis, and absent or dysplastic renal parenchyma.
Renal scintigraphy confirms the diagnosis of multicystic dysplastic kidney by showing absence of function. It should be noted that severe hydronephrosis and the multicystic dysplastic kidney may be indistinguishable by scintigraphy.
 Figure 8.20 Neonatal autosomal recessive polycystic disease. A. Prone longitudinal scan reveals an enlarged, echogenic right kidney (cursors) and loss of corticomedullary differentiation with tiny cysts. B. Transverse scan of the opposite kidney shows multiple small cysts, echogenic cortex, and no corticomedullary differentiation. C. Transverse sonogram of the liver shows dilated intrahepatic ducts (arrowheads). |
Multicystic dysplastic kidney is treated nonoperatively, because the natural history is spontaneous decrease in size during the first year of life (12). Surgery is performed if there is respiratory distress because of thoracic compression by the large cystic kidney. Patients treated conservatively are followed with serial sonograms, because of a possible increased risk of Wilms tumor.
BILATERAL CYSTIC DISEASE
Autosomal Recessive Polycystic Disease
Polycystic renal disease can be classified into autosomal recessive and autosomal dominant types. In the autosomal recessive form of disease, previously known as infantile polycystic kidney disease, the kidneys are markedly enlarged but retain their reniform shape. Histologic studies show that these cysts are predominantly dilated collecting tubules, which are radially arranged, extending from the medulla to the subcapsular cortex. The nephrons are normal or minimally altered. Virtually all patients with autosomal recessive cystic disease have hepatic abnormalities, characterized by biliary ductal ectasia and periportal fibrosis. The clinical features of autosomal recessive polycystic disease are dependent on the age of presentation. Infants tend to have more severe renal disease and milder hepatic disease. Patients who survive the neonatal period and come to clinical attention later in life have milder renal disease and more severe hepatic disease (usually portal hypertension).
The clinical features of autosomal recessive polycystic disease are dependent on the age of presentation.
In the neonate, sonography typically shows bilateral renal enlargement, diffusely increased parenchymal echogenicity, and poor corticomedullary differentiation (13) (Fig. 8.20). Occasionally, there may be a thin rim of hypoechoic parenchyma at the periphery that is thought to be compressed cortex. Cortical cysts also may be evident. On CT, the kidneys are enlarged and show a striated pattern of contrast material excretion, representing contrast stasis in dilated tubules (Fig. 8.21). The renal parenchyma is hypointense on T1-weighted MR images and hyperintense on T2-weighted images. Dilated bile ducts may be seen on sonography, CT, and MRI.
The kidneys in older children may be normal-sized or enlarged (13) (Fig. 8.22). The echogenicity is increased, but it often is limited to the medullary area. There usually is some parenchymal striation on CT but less than that in the neonate. Nodular hepatic margins, splenomegaly, and esophageal varices, typical of cirrhosis, are other common findings.
Autosomal Dominant Polycystic Disease
Autosomal dominant polycystic disease, previously known as adult polycystic renal disease, is a hereditary disorder resulting from an abnormality on the short arm of chromosome 16. Pathologically, multiple cysts of varying size are present in the cortex and medulla with islands of normal parenchyma interspersed between the cystic areas. The cysts communicate with the nephrons as well as the collecting tubules. Cysts are also common in the liver, but there usually is no significant periportal hepatic fibrosis. Cysts also may be found in the pancreas, lungs, spleen, ovaries, testes, and seminal vesicles. Clinical findings include palpable abdominal masses in neonates and hypertension, flank pain, and hematuria in children and adolescents.
In the neonate, sonography demonstrates enlarged, diffusely echogenic kidneys. The sonographic findings are similar to those of autosomal recessive polycystic disease, and the diagnosis needs to be based on family history or renal biopsy.
In the neonate, sonographic findings of autosomal dominant and recessive polycystic disease are similar.
In older children and adolescents, the kidneys may be of normal size or enlarged and have lobulated or smooth borders. Multiple cysts are seen throughout the full thickness of the renal parenchyma. They have the typical appearance of simple cysts on sonography, CT, and MRI (Fig. 8.23).
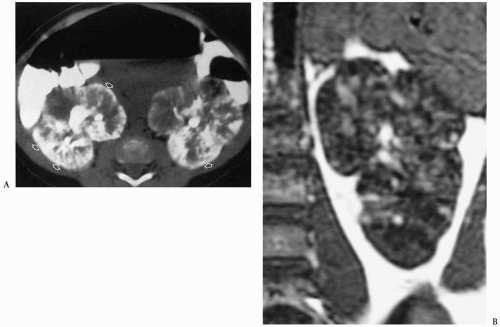 Figure 8.21 Neonatal autosomal recessive polycystic disease. A. CT scan demonstrates prolonged cortical enhancement (open arrows) and contrast-filled tubules, creating a striated appearance. (From Siegel MJ, ed. Pediatric body CT. Philadelphia: Lippincott Williams & Wilkins, 1999, with permission.) B. Coronal T1-weighted MR image in another image shows small cysts replacing normal parenchyma. |
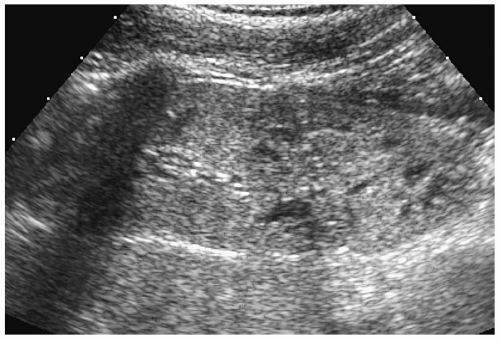 Figure 8.22 Childhood autosomal recessive polycystic disease. Longitudinal sonogram of a 7-year-old girl shows a normalsize right kidney with mildly echogenic parenchyma and loss of corticomedullary differentiation. |
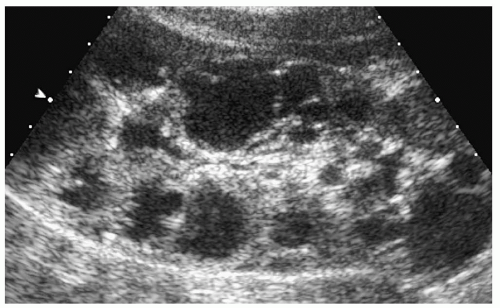 Figure 8.23 Autosomal dominant polycystic disease. Longitudinal sonogram from an 8-year-old girl with a family history of renal cysts shows numerous cortical cysts of various sizes. |
 Figure 8.24 Glomerulocystic disease in a neonate with bilateral flank masses. Sagittal sonogram in a neonate shows an enlarged, echogenic right kidney (K) containing multiple cysts of varying sizes. The sonographic appearance is similar to that of autosomal recessive and autosomal dominant polycystic disease in the neonate (L, liver). (Image courtesy of D. Gregory Bates, M.D., Columbus, Ohio.) |
Glomerulocystic Disease
Glomerulocystic disease is a rare sporadic renal disorder characterized by cystic dilatation of the Bowman space, with a variable degree of dilatation of the proximal collecting tubules. Periportal hepatic fibrosis, bile duct hyperplasia and dilatation, and hepatic cysts also can be seen. Sonographic findings are similar to those of autosomal recessive and dominant polycystic disease in the neonate (Fig. 8.24). The diagnosis is usually established by renal biopsy.
Cystic Disease of the Renal Medulla
Medullary sponge kidney, also known as medullary tubular ectasia, is a sporadic rather than an inherited disorder. Pathologically, there is cystic dilatation of the collecting tubules in the area of the renal pyramids. The condition is asymptomatic, unless complications, such as infection or urolithiasis, occur. Uncomplicated tubular ectasia is difficult to recognize on imaging studies. Urolithiasis is seen as hyperechoic renal pyramids on sonography and as small calcifications on CT (see section on calcifications).
Juvenile nephronophthisis and medullary cystic disease are hereditary diseases characterized by polyuria, polydipsia, salt-wasting, severe anemia, and ultimately renal failure. Pathologically, the kidneys are small or normal-sized, and cysts are present in the medulla and at the corticomedullary junction. Juvenile nephronophthisis is inherited as an autosomal recessive trait and becomes symptomatic in childhood or adolescence. Medullary cystic disease is inherited as an autosomal dominant trait and usually becomes symptomatic in adulthood. Imaging studies show small kidneys, loss of corticomedullary differentiation, and a variable number of medullary or corticomedullary cysts (Fig. 8.25).
Juvenile nephronophthisis and medullary cystic disease are characterized by small kidneys, loss of corticomedullary differentiation, and medullary or corticomedullary cysts.
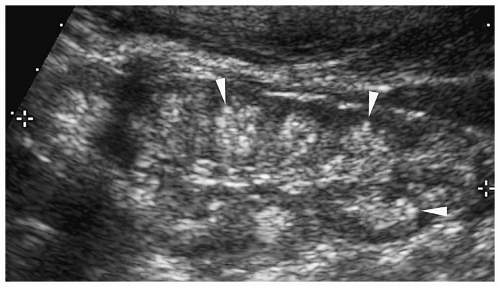 Figure 8.25 Medullary cystic disease. Longitudinal sonogram in a child with hematuria shows a small right kidney (cursors) with thinned cortex. Tiny echogenicities (arrowheads) are noted at the corticomedullary junction, arising from numerous tiny cysts. The left kidney had a similar appearance. |
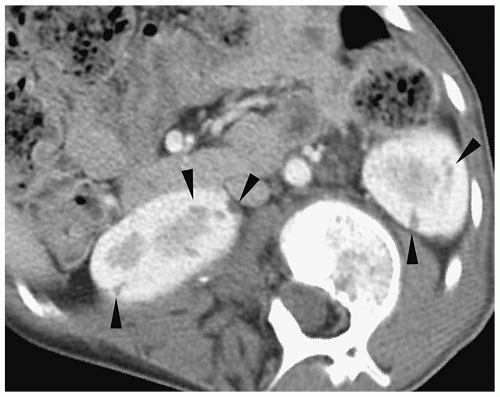 Figure 8.26 Tuberous sclerosis. Contrast-enhanced CT in a teenager with severe scoliosis scan shows small cortical cysts throughout both kidneys (arrowheads). |
Renal Cysts Associated with Multisystem Disorders
Tuberous sclerosis is an autosomal dominant disorder resulting from a defect on chromosome 9. Classic clinical features are seizures, mental retardation, and cutaneous lesions. Von Hippel-Lindau disease is due to an error on the short arm of chromosome 3. Retinal and central nervous system hemangioblastomas, pancreatic cysts and islet cell tumors, and pheochromocytoma are common clinical findings. Renal cysts are common in both disorders, and they are usually bilateral (Fig. 8.26). Correlation with family history and clinical findings usually permits the correct diagnosis to be established.
Renal cysts are common in tuberous sclerosis and Von Hippel-Lindau disease.
Cysts can also be found in a variety of other syndromes, including Turner syndrome, Meckel syndrome (microcephaly, polydactyly, posterior encephalocele), Jeune asphyxiating thoracic dystrophy (small chest, respiratory failure, renal dysplasia), oral-facial-digital syndrome, Zelweger syndrome (cerebrohepatorenal syndrome), and Beckwith-Wiedemann syndrome. Their imaging characteristics are identical to those of simple cysts. Correlation with clinical findings usually permits the correct diagnosis.
RENAL NEOPLASMS
Because of its high sensitivity and ease of performance, ultrasonography is usually the initial imaging examination in an infant or child with a palpable abdominal mass. If the sonogram shows a benign condition such as a hydronephrotic kidney or renal cystic disease, renal scintigraphy can be performed to evaluate function or to confirm the diagnosis. If the mass is solid and a malignant neoplasm is suspected, CT or MR imaging is performed to better delineate the extent of the disease (2,3,4).
MALIGNANT TUMORS IN YOUNGER PATIENTS
Wilms Tumor
Wilms tumor (nephroblastoma) is the most common renal malignancy in children (14,15,16). Affected children usually present before 5 years of age (mean age, 3 years). The common clinical finding is a palpable abdominal mass. Other findings include abdominal pain, fever, microscopic or gross hematuria, and hypertension. Occasionally, patients present with the Budd-Chiari syndrome secondary to hepatic vein invasion by tumor. Associated abnormalities, such as Beckwith-Wiedemann syndrome, nonfamilial aniridia, congenital hemihypertrophy, and Drash syndrome (male pseudohermaphrodism and renal failure)
occasionally prompt evaluation. Approximately, 10% of children have metastatic disease at presentation. Metastases are characteristically to the lungs (10% to 20%) and less frequently to the liver. Bilateral synchronous tumors occur in 5% to 10% of patients. Wilms tumor may occur in extrarenal sites, such as the retroperitoneum, inguinal region, pelvis, or thorax. These tumors are not usually associated with a primary renal tumor.
occasionally prompt evaluation. Approximately, 10% of children have metastatic disease at presentation. Metastases are characteristically to the lungs (10% to 20%) and less frequently to the liver. Bilateral synchronous tumors occur in 5% to 10% of patients. Wilms tumor may occur in extrarenal sites, such as the retroperitoneum, inguinal region, pelvis, or thorax. These tumors are not usually associated with a primary renal tumor.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


