Fig. 16.1
Normal infant adrenal gland – US. Longitudinal ultrasound image of the right adrenal gland (a) and transverse ultrasound image of the left adrenal gland (b) in a 2-month-old girl illustrate the normal appearance (arrows) with hypoechoic adrenal cortex surrounding the central echogenic adrenal medulla creating an “Oreo cookie” appearance
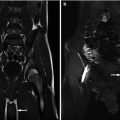
The adrenal glands can be visualized anteromedial to the upper pole of the ipsilateral kidney at all ages on both CT and MR [1]. The right adrenal gland lies medial to the right lobe of the liver, lateral to the right crus of the diaphragm, and posterior to the inferior vena cava [1]. The left adrenal gland lies medial to the spleen, lateral to the aorta and left crus of the diaphragm, and posterior to the pancreatic tail and stomach [1]. To estimate normal adrenal size, the adrenal glands should be thinner than the adjacent diaphragmatic crura on axial images [1]. CT and MR can similarly demonstrate detailed information on adrenal anatomy; though, in determining the modality of choice, their respective risks and benefits should be considered (e.g., radiation exposure from CT and the risk of sedation during MR). On CT, the adrenal gland’s soft tissue attenuation is similar to the liver (Fig. 16.2) [11]. For MR, on spin-echo T1-weighted images, adrenal glands have intermediate signal intensity (less than fat and similar to the liver); on T2-weighted and fat-suppressed images, adrenal glands are much brighter than fat and slightly brighter than the liver (Fig. 16.3) [11].
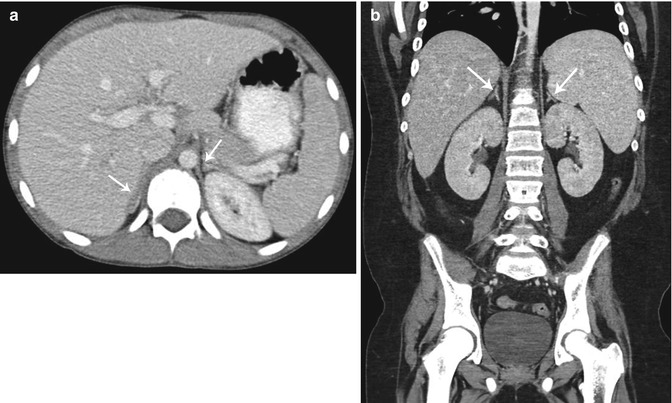
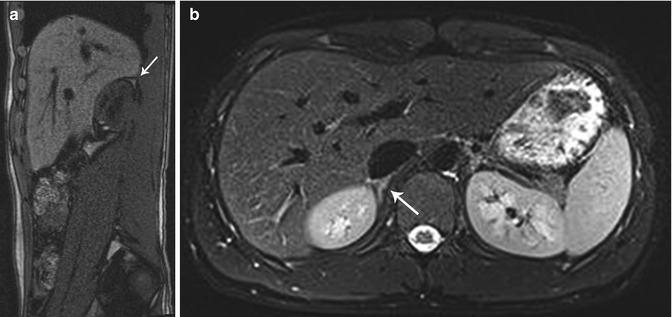
Fig. 16.2
Normal adrenal gland – CT. Axial contrast-enhanced CT image in a 12-year-old girl (a) and coronal contrast-enhanced CT image in a 14-year-old boy (b) show normal size and appearance of the adrenal glands (arrows) which are isodense to the liver
Fig. 16.3
Normal adrenal gland – MR. MR images in a 13-year-old boy reveal normal adrenal gland. On sagittal T1 images (a) the adrenal gland (arrows) is intermediate signal intensity similar to the liver. On axial T2 fat-suppressed images (b) the right adrenal gland (arrow) is intermediate signal intensity and brighter than the adjacent liver
Anomalies of Shape and Position of the Adrenal Gland
As adrenal development and renal development are separate processes, adrenal glands will develop in their normal position within the retroperitoneum despite ipsilateral renal agenesis, malrotation, or ectopia [12]. However, in these cases, they are often flattened or discoid in shape, as well as slightly longer and thicker (referred to as a “straight adrenal gland”) (Fig. 16.4) [5, 6, 11, 13]. This straight adrenal gland is not seen after nephrectomy or acquired renal atrophy [5].
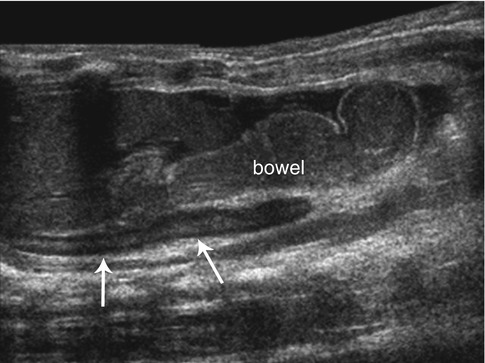
Fig. 16.4
Renal agenesis. Longitudinal ultrasound image of the left upper quadrant in a neonate with left renal agenesis. Bowel loops fill the left renal fossa with a straight, discoid left adrenal gland (arrows) seen
Anomalies of the adrenal glands are exceedingly rare. The two most common fusion anomalies are the circumrenal adrenal and the horseshoe adrenal [3, 14]. In the circumrenal adrenal, the fused limbs fuse around the upper pole of the ipsilateral kidney [3, 14]. Horseshoe adrenal describes fusion of the right and left adrenal glands in the midline anterior to the spine and posterior to the aorta [3, 14]. Horseshoe adrenals are often associated with renal anomalies (horseshoe kidney, renal agenesis), central nervous system anomalies such as neural tube defects, and asplenia with visceral heterotaxy [3, 14]. The isthmus of the horseshoe adrenal usually passes posterior to the aorta, but in association with asplenia the isthmus passes anterior [14].
Adrenal rests, or accessory adrenal glands, are mainly composed of cortical tissue, with the occasional presence of medullary tissue. They can be found anywhere along the path of gonadal descent in the retroperitoneum. Most commonly they are located near the celiac plexus, but can also be seen along the course of the gonadal veins and within the broad ligament, ovary, inguinal canal, testes, and epididymis [3]. Adrenal rests typically atrophy with time; therefore, although they can be found in up to 50 % of neonates, they are found in only 1 % of adults [7]. Males with congenital adrenal hyperplasia have a high prevalence of persistent adrenal rests in the testes [15].
Anomalies of Size of the Adrenal Gland
Congenital Adrenal Hyperplasia
Congenital adrenal hyperplasia (CAH) is collection of autosomal recessive disorders characterized by low cortisol production, potential aldosterone deficiency, and androgen excess due to an enzymatic defect in the cholesterol-steroid biosynthesis pathway [5]. Over 95 % of cases are due to a deficiency in the enzyme 21-hydroxylase [5]. Androgen excess leads to virilized genitalia in female infants and early virilization in male infants [11]. In the most severe form, concomitant aldosterone deficiency leads a salt-losing crisis in either sex during the newborn period [5]. Diagnosis can be made by a very high concentration of 17-hydroxyprogesterone (17-OHP) after three days of life [16].
Most neonates with CAH have enlargement of the adrenal glands with a width measurement of >4 mm and a length measurement of >20 mm [17]. Additional signs which may be seen on US include a cerebriform appearance of the surface of the adrenal gland and a stippled central adrenal echogenicity [18, 19]. In a series by Al-Alwan et al., US in the immediate neonatal period had a sensitivity of 92 % and a specificity of 100 % for the diagnosis of CAH and may be employed before conclusive 17-OHP levels are available [18]. Diagnosis of CAH can be made by the demonstration of two of three sonographic signs: (1) adrenal limb width of >4 mm, (2) cerebriform or crenated appearance of the surface of the adrenal gland, and (3) replacement of the central hyperechoic stripe with a diffusely stippled pattern of echogenicity or a diffuse thickened band of echogenicity (Fig. 16.5) [18].
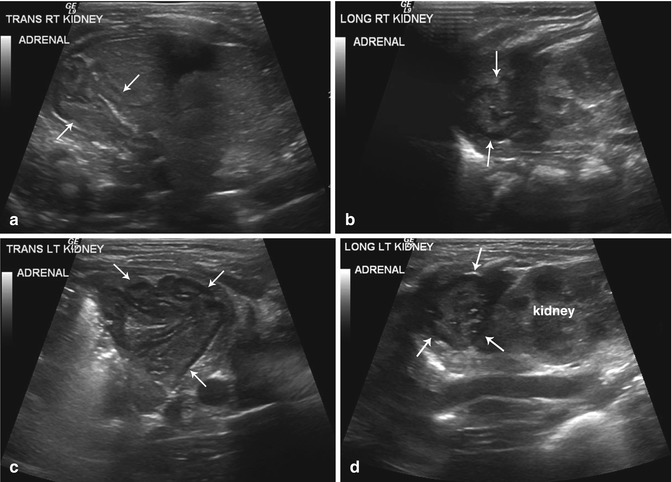
Fig. 16.5
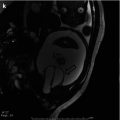
Congenital adrenal hyperplasia. Retroperitoneal ultrasound images from both flanks (a–d) performed on a newborn girl with ambiguous genitalia due to congenital adrenal hyperplasia reveal enlarged adrenal glands with a cerebriform contour (arrows)
Case reports suggest that CAH due to deficiencies in 11β-hydroxylase or 3β-hydroxysteroid dehydrogenase leads to similar changes on adrenal US [20]. Lipoid adrenal hyperplasia is a rare form of CAH due to cholesterol desmolase deficiency, and adrenal glands appear enlarged with echogenicity or attenuation similar to fat due to accumulation of cholesterol and its esters [21].
Adrenal Hyperplasia
When adrenocortical hyperplasia presents in older children, it is classified as primary or secondary [1]. Primary adrenocortical hyperplasia results in either Cushing syndrome or, less commonly, primary hyperaldosteronism (Conn syndrome) [1]. Secondary adrenocortical hyperplasia is due to excess ACTH: either endogenous in those with Cushing disease or ectopic ACTH production or exogenous in those receiving ACTH administration (e.g., for infantile spasms) [1]. Clinical features of Cushing syndrome include central obesity, moon facies, buffalo hump, proximal muscle weakness, easy bruisability, abdominal striae, hypertension, dyslipidemia, insulin resistance, and elevated 24-h urinary cortisol and 17-hydroxycorticoids [22]. Clinical manifestations of Conn syndrome include muscle weakness, hypokalemia, and hypertension [23].
Adrenal glands are not normally visible by US in older children; therefore, if they are visible, the diagnosis of adrenal hyperplasia should be entertained [1]. CT and MR may be used to visualize the adrenals in older children with adrenal hyperplasia. Hyperplastic adrenal glands in these cases will be bilaterally, symmetrically, and evenly enlarged with increased relative enhancement [1]. Alternatively, in some cases of adrenal hyperplasia, adrenal glands may be normal in size, demonstrate uneven enlargement, or contain small nodular areas (Fig. 16.6) [1].
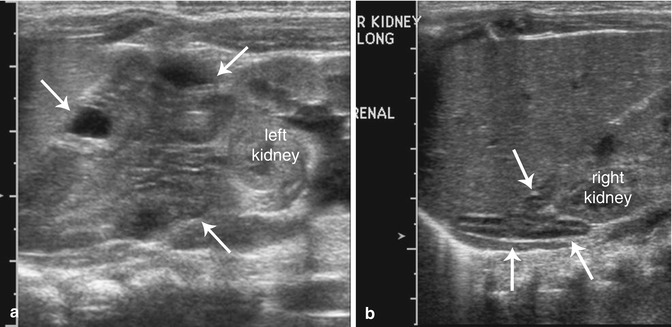
Fig. 16.6
Atypical congenital hyperplasia. US images from the left flank (a) reveal an enlarged adrenal gland with a cerebriform contour (arrows) and an anatomically normal-appearing adrenal gland from the right flank (b)
Cushing syndrome and Conn syndrome can also be associated with adrenal adenomas. Benign adrenal adenomas cannot be reliably distinguished from adrenal carcinomas based on histopathology, clinical features, or imaging [24]. However, in general, adrenal adenomas are smaller (<6 cm) compared to adrenal carcinoma and have less heterogeneity on US, CT, and MR. Adrenal carcinomas can have a similar appearance to adrenal adenomas, but tend to be larger and more complex [24]. The only definitive signs of malignancy are hematogenous metastases or venous spread [24].
Primary pigmented nodular adrenocortical disease is a rare form of Cushing syndrome and is associated with Carney complex (Sertoli cell tumors of the testis, cardiac myxomas, soft tissue myxomas, and skin pigmentation) [25]. On imaging, the adrenal gland has multiple, <2 mm cortical-secreting adenomas with atrophic cortex between the nodules [25].
Wolman Disease
Wolman disease, a rare disorder, is an inherited deficiency of lysosomal acid lipase leading to the accumulation of cholesterol esters and triglycerides in many organs, especially in the adrenals [3]. Wolman disease presents in the first few weeks of life with hepatosplenomegaly with abdominal distension, jaundice, vomiting, diarrhea, steatorrhea, anemia, and growth failure and is rapidly progressive leading to death in the first year [3, 5]. On US, the adrenals appear markedly enlarged with calcifications appearing as a long, linear echogenic band with posterior acoustic shadowing [5]. On CT, the adrenals also appear enlarged with a cortical distribution of calcification [5]. Plain film will also demonstrate the densely calcified adrenal gland (Fig. 16.7) [5]. Imaging of adrenals in Wolman disease can look similar to resolving adrenal hemorrhage; however, in adrenal hemorrhage the adrenals are smaller and have globular calcifications [5, 26, 27].
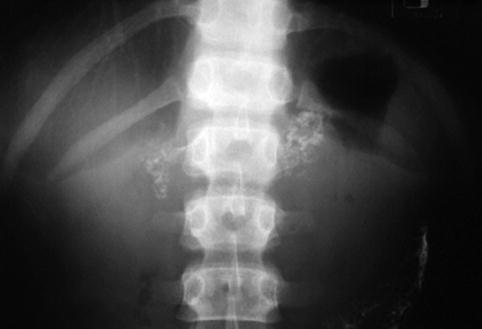
Fig. 16.7
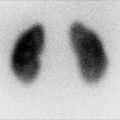
Wolman disease. Abdominal radiograph in a 6-month-old boy reveals calcifications outlining the enlarged adrenal glands, findings typical of Wolman disease
Adrenal Masses
Adrenal masses in children and neonates may be attributable to hemorrhage, neoplasms, cysts, or abscesses [5]. The age and clinical presentation of the child, in conjunction with the imaging features of the mass, will allow one to develop an appropriate list of diagnostic considerations.
In the neonate with an adrenal mass lesion, the most likely entities include adrenal hemorrhage, neuroblastoma, and rarely extralobar pulmonary sequestration. In children less than 5 years of age, neural crest tumors including neuroblastoma and ganglioneuroblastoma are more likely than adrenal hemorrhage and adrenocortical neoplasms except when the child is exhibiting signs and symptoms of a hormonally active tumor. In older children and adolescents, adrenal masses may be related to neural crest tumors (more frequently the mature ganglioneuromas as opposed to neuroblastoma or ganglioneuroblastoma), as well as other tumors including pheochromocytomas and adrenocortical neoplasms. Other rarely seen adrenal masses in children include rhabdoid tumors, myelolipomas, and smooth muscle adrenal tumors [28].
Adrenal Hemorrhage
Adrenal hemorrhage in the perinatal period can occur in response to perinatal stress (such as difficult or traumatic delivery, hypoxia, or sepsis) or bleeding disorders [29]. Large babies such as those of diabetic mothers or with Beckwith-Wiedemann syndrome have a higher predisposition [30]. Associated clinical signs may include palpable flank mass, anemia, jaundice, or rarely hypovolemic shock. However, there is normally no associated adrenal insufficiency in the immediate phase or long-term as the major insult is to the regressing fetal cortex [5]. Beyond the neonatal period, adrenal hemorrhage is frequently seen in the setting of trauma but has also been documented to occur in older children with overwhelming sepsis (specifically Neisseria meningitidis), steroid therapy, anticoagulation therapy, and after liver transplantation [5, 11].
Adrenal hemorrhage more often occurs on the right (70 %), and in only 10 % of cases does it occur bilaterally [5]. Hemorrhage size can vary from as small as a few centimeters and up to several centimeters [5]. With conservative management, the natural course of adrenal hemorrhage is central liquefaction with resorption of blood leading to eventual decrease in size to the normal shape and a residual focus of calcification [5].
US is the modality of choice for initial imaging of adrenal masses in neonates as well as for follow-up assessment [31, 32]. On initial US, adrenal hemorrhages have a varied appearance depending on the duration of the hemorrhage. They usually have complex echogenicity with echogenic and echo-free areas but can also appear evenly echogenic, hypoechoic, or anechoic [5]. Smaller hemorrhages may be focal or retain the shape of the adrenal (triangular or crescent-shaped), and adjacent normal adrenal tissue can be identified. Larger adrenal hemorrhages are usually round in shape, and normal unaffected adrenal tissue can be hard to identify (Figs. 16.8a, b) [5]. Large hemorrhages may encompass the upper pole of the kidney (with the appearance of a perinephric hemorrhage) and may track down the retroperitoneum. Clinically, this can lead to scrotal swelling and hematoma, which may mimic testicular torsion [5, 33]. Occasionally, there may be associated ipsilateral renal vein thrombosis, especially on the left since the adrenal vein drains into the left renal vein [5]. To help differentiate adrenal hemorrhage (which is avascular) from neuroblastoma or other adrenal neoplasms, evaluation with color and spectral Doppler assessment should be performed (Fig. 16.8c). Additionally, ultrasound examination should include evaluation of the liver to assess for the presence of hepatic metastases and evaluation of the remainder of the abdomen and pelvis to look for metastatic lymphadenopathy, which can be seen in association with adrenal tumors.
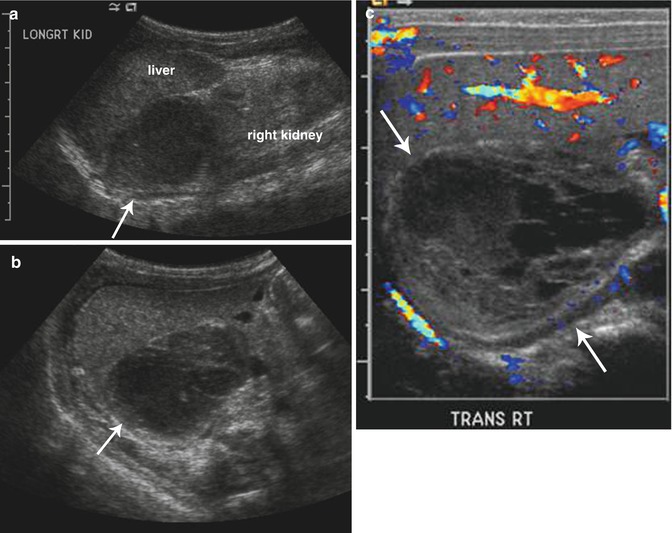
Fig. 16.8
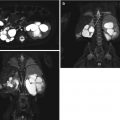
Adrenal hemorrhage. Longitudinal (a) and transverse (b) ultrasound images in a 2-week-old boy with right adrenal hemorrhage reveal a rounded heterogeneously hypoechoic right adrenal mass (arrow). Color Doppler ultrasound image (c) shows that the right adrenal mass is avascular (arrows) and suggestive of an adrenal hemorrhage
Because of the varied imaging characteristics of adrenal hemorrhage in the acute phase, it may be difficult initially to distinguish from neuroblastoma [34, 35]. Thus, follow-up US imaging is of utmost importance in differentiating a resolving adrenal hemorrhage from a neuroblastoma. Masses due to adrenal hemorrhage decrease in size over several weeks as the hemorrhage liquefies and resorbs and become more hypoechoic or anechoic (Fig. 16.9) [36], whereas in contrast, the size of a neuroblastoma is unlikely to decrease. In cases where the diagnosis is uncertain, a short delay with serial US imaging is not harmful as neonatal neuroblastoma has a relatively good prognosis [11].
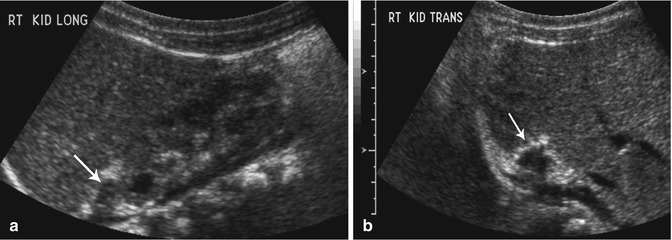
Fig. 16.9
Resolving adrenal hemorrhage. Follow-up longitudinal (a) and transverse (b) ultrasound images (obtained 2 weeks after the images shown in Fig. 16.8) reveal diminution in the size of the right adrenal gland (arrows) and increased peripheral echogenicity (likely early calcification) consistent with a resolving adrenal hemorrhage
In some instances adrenal calcification may be incidentally seen on imaging (plain film, CT, MR) on older children. If the calcification is confined to adrenals of normal size and without evidence for a soft tissue mass, it is assumed to have resulted from previous adrenal hemorrhage in the neonatal period and is of no clinical significance (Fig. 16.10) [5].
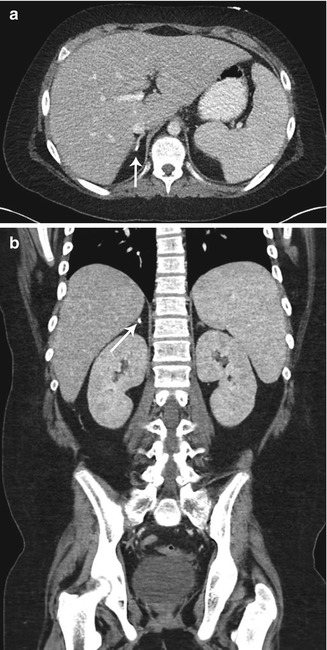
Fig. 16.10
Old adrenal hemorrhage. Axial (a) and coronal (b) contrast-enhanced CT images of the abdomen in a 10-year-old boy reveal a small focus of calcification in the right adrenal gland (arrows) with no associated soft tissue mass, presumed the sequela of prior adrenal hemorrhage
In neonates with a suprarenal retroperitoneal mass, the possibility of an intra-abdominal extralobar pulmonary sequestration must also be considered in addition to adrenal hemorrhage and neuroblastoma [3]. Intra-abdominal extralobar pulmonary sequestrations occur more commonly on the left than the right, are usually hyperechoic, and can contain cysts related to coexistent congenital pulmonary airway malformation (Fig. 16.11) [28, 37]. As imaging with US, CT, and MR is rarely diagnostic, surgical diagnosis and treatment is often necessary [38].
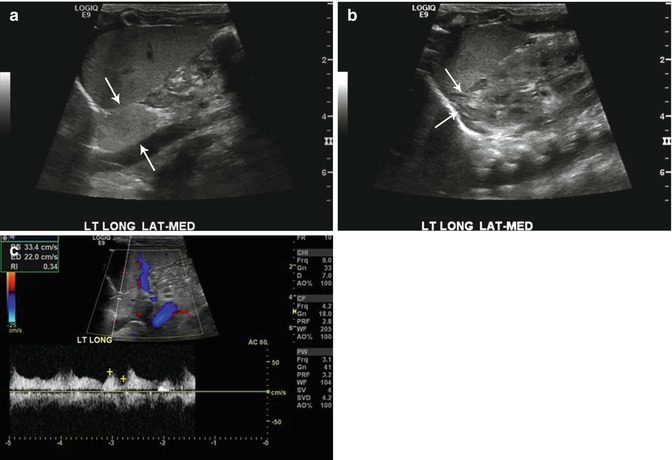
Fig. 16.11
Retroperitoneal extralobar pulmonary sequestration. Longitudinal ultrasound images of the left upper abdomen (a) in an 11-day-old boy with prenatally diagnosed mass reveal an echogenic mass medially (arrows) and a normal-appearing adrenal gland laterally (b). Doppler assessment confirmed internal blood flow (c) with large systemic artery typical for an extralobar pulmonary sequestration
Medullary Neoplasms
Medullary adrenal neoplasms include the neuroblastoma, ganglioneuroblastoma, ganglioneuroma, and pheochromocytoma. These can occur within the medulla of the adrenal gland but also along the sympathetic nervous chain [1].
Neuroblastoma
Neuroblastoma, ganglioneuroblastoma, and ganglioneuroma are tumors which arise from the neural crest cells of the sympathetic nervous system [39]. Neuroblastomas are malignant tumors composed of immature neuroblasts. Ganglioneuroblastomas are composed of both immature and mature cells and have malignant potential. Ganglioneuromas composed entirely of mature gangliocytes and mature stroma are benign [39].
Neuroblastoma represents 8–10 % of all childhood cancers making it the most common extracranial solid tumor of childhood [39–41]. The median age of neuroblastoma diagnosis is 19 months, most present between 1 and 5 years of age [3]. Most primary tumors are in the abdomen (65 %), although children have a higher frequency of adrenal tumors than infants (40 % vs. 25 %). The remaining abdominal and pelvic tumors mostly originate in the paravertebral sympathetic ganglia or in the presacral area from the organ of Zuckerkandl [3].
Most children present with abdominal pain or a palpable mass, but others are identified by manifestations of their metastatic disease, as up to 70 % of patients have metastases at presentation [3]. Patterns of metastases vary with age at presentation, and locations can include lymph nodes, liver, skeleton, bone marrow, and skin [3]. Neonates and younger infants more commonly have cutaneous lesions (blueberry muffin syndrome) and extensive hepatic involvement (however, hepatic metastases can occur at any age), whereas older infants and children more commonly have skeletal metastases [3]. Additionally, patients may present with paraneoplastic syndromes. Serum or urinary levels of catecholamines or their metabolites (vanillylmandelic acid (VMA), homovanillic acid (HVA)) are increased in 90 % of children with neuroblastoma [42]; however, secretion of catecholamines rarely leads to symptoms such as those seen in pheochromocytoma: paroxysmal hypertension, palpitations, flushing, and headaches. Secretion of catecholamines or vasoactive intestinal peptide (VIP) may lead to severe watery diarrhea, hypokalemia, and acidosis [43]. Additionally, one can present with acute myoclonic encephalopathy comprised of myoclonus, opsoclonus (rapid multidirectional eye movements), and cerebellar ataxia; this is thought to be due to an immune response to the primary tumor leading to production of anti-neural antibodies that cross-react with cerebellar tissue [44].
The prognosis of neuroblastoma is related to age, stage at presentation (see Table 16.1), and tumor site [1]. According to the International Neuroblastoma Staging System (INSS), the distribution at presentation is as follows: stage 1, 17 %; stage 2A/2B, 16 %; stage 3, 16 %; stage 4, 44 %; and stage 4S, 7 % [41]. However, newer staging systems based on imaging, instead of surgical, findings are in development [46]. Favorable prognosis occurs in patients <1 year of age, low-stage disease at presentation, and tumors arising from extra-abdominal sites. For example, 2-year survival is 80 % in patients with localized disease, whereas it is less than 5 % for patients with skeletal metastases [11]. Additionally, poor prognostic signs are N-myc amplification (>10 copies), allelic loss of chromosome 1p, and diploid karyotype, whereas favorable prognostic factors are unamplified N-myc oncogene, absence of abnormalities on chromosome 1p, triploid karyotype, and well-differentiated stroma on histology [39].
Table 16.1
International neuroblastoma staging system
Stage | Description |
|---|---|
1 | Localized tumor with complete gross excision, with or without microscopic residual disease; representative ipsilateral lymph nodes negative for tumor microscopically |
2A | Localized tumor with incomplete gross excision; representative ipsilateral nonadherent lymph nodes negative for tumor microscopically |
2B | Localized tumor with or without complete gross excision, with ipsilateral nonadherent lymph nodes positive for tumor. Enlarged contralateral lymph nodes must be negative microscopically |
3 | Unresectable unilateral tumor infiltrating across the midline, with or without regional lymph node involvement; localized unilateral tumor with contralateral regional lymph node involvement; or midline tumor with bilateral extension by infiltration (unresectable) or by lymph node involvement. The midline is defined as the vertebral column. Tumors originating on one side and crossing the midline must infiltrate to or beyond the opposite side of the vertebral column |
4 | Any primary tumor with dissemination to distant lymph nodes, bone, bone marrow, liver, skin, and/or other organs, except as defined for stage 4S |
4S | Localized primary tumor, as defined for stage 1, 2A, or 2B, with dissemination limited to skin, liver, and/or bone marrow (limited to infants younger than 1 year). Marrow involvement should be minimal (i.e., <10 % of total nucleated cells identified as malignant by bone biopsy or by bone marrow aspirate). More extensive bone marrow involvement would be considered stage 4 disease. The results of the metaiodobenzylguanidine (MIBG) scan, if performed, should be negative for disease in the bone marrow |
Treatment consists of a combination of surgery, chemotherapy, and radiation depending on the stage at presentation. Primary surgical resection is used for more localized tumors and chemotherapy for unresectable lesions or in a neoadjuvant setting to shrink lesions sufficiently for delayed surgical resection [39].
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


