Central Nervous System Manifestations of the Phakomatoses
Susan I. Blaser
James G. Smirniotopoulos
Frances M. Murphy
The neurocutaneous syndromes, commonly called the phakomatoses, are the usual and most extensively studied of the more commonly encountered inherited genetic central nervous system (CNS) syndromes. These disorders have variable patterns of inheritance and commonly arise from new spontaneous mutations. While not all of the neurocutaneous syndromes have a known mutation, once a mutation is present in an individual, it may be inherited by the next generation.
Neuroimaging plays a critical and central role in the diagnosis, staging, and management of patients with these disorders and in the appropriate testing and counseling of first-degree relatives (parents, siblings, and children). Because these diseases have prominent and often pathognomonic imaging findings, the diagnosis can be suggested or be made entirely on the basis of neuroimaging, helping to direct the genetic testing. Imaging and clinical findings may develop over time, so individuals with these syndromes usually require routine periodic clinical and neuroimaging surveillance to monitor the progression of existing lesions and/or the development of new manifestations.
TABLE 5.1 Most Frequent Neurocutaneous Syndromes | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
General Concepts
The terms “neurocutaneous” and “phakomatosis” imply that the described condition affects both the skin and the CNS. The most frequent and important of the 30 or more neurocutaneous syndromes are listed in Table 5.1.
The Dutch ophthalmologist Jan van der Hoeve coined the word “phakoma” to describe the ocular/retinal findings of von Recklinghausen neurofibromatosis type 1 (NF1) and tuberous sclerosis (TS). He subsequently added Sturge–Weber syndrome and von Hippel–Lindau syndrome (VHL) to his grouping of phakomatoses. While these diseases are grouped together as neurocutaneous syndromes, some—for example, VHL—do not actually affect the skin at all. Through the years, additional rare neurocutaneous syndromes, such as PHACES (posterior fossa malformations, hemangiomas, arterial anomalies, coarctation of the aorta, cardiac defects, eye abnormalities, defects of the sternum, and abdominal raphe) and neurocutaneous melanosis, have been described. Recent advances in molecular biology, including linkage analysis, polymerase chain reaction, and DNA sequencing, have been able to localize the chromosomes, genes, and in some cases even the protein products related to many of the phakomatoses (Table 5.2).
TABLE 5.2 Phakomatoses: Genes | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
The Common Phakomatoses
Neurofibromatosis Type 1
The National Institutes of Health (NIH), through its consensus panels, has defined two distinct inherited nerve sheath disorders, NF1 and neurofibromatosis type 2 (NF2) (1). There are as many as eight different subtypes of neurofibromatosis. NF1 is the classic “peripheral” disease described by the German pathologist Friedrich Daniel von Recklinghausen. Affected individuals may have very prominent, clinically evident external lesions, including superficial tumors (neurofibromas) and macular hyperpigmentation (café-au-lait spots) that facilitate clinical recognition. NF1 is characterized by an inherited predisposition for the development of benign peripheral nerve sheath tumors (neurofibromas) and significant CNS abnormalities, including true neoplasms (usually optic nerve and visual pathway gliomas [ONGs]) and dysplastic and hamartomatous/heterotopic lesions. Many of the more common manifestations and diagnostic criteria of NF1 are summarized in Table 5.3 (1). DNA analysis testing may be helpful in patients suspected of having NF1. Of historical interest, Joseph Merrick (the original “Elephant Man”) is now felt to have suffered, not from neurofibromatosis, but from proteus syndrome. While genetic testing is possible in each of these neurocutaneous disorders, DNA confirmation was not possible in his case due to preservation and storage issues involving bone and skin samples (2).
NF1, the most common of the neurocutaneous syndromes, is an autosomal dominant disorder without racial or sexual predilection. The incidence is estimated at 1 in 3,000 to 1 in 4,000 persons (3). NF1 is an extremely pleomorphic disorder. Individuals with classic stigmata may suffer significant morbidity and mortality from symptomatic lesions or be asymptomatic, with few obvious findings and a normal life span. Asymptomatic subjects may not be diagnosed until an autopsy or diagnostic testing for an unrelated disease reveals the characteristic features or until affected offspring are born. However, in most individuals, the diagnosis is commonly made on the basis of the prominent cutaneous findings or is detected while screening first-degree relatives of an affected individual.
Less than half of the patients with NF1 have a positive family history, suggesting a high rate of spontaneous mutation. The lack of positive family history in older literature may reflect the absence at the time of a thorough clinical and neuroimaging evaluation and genetic testing. Genetic linkage analyses have shown that the gene locus for NF1 is at 17q11.2 (4,5). The normal gene appears to function as a tumor-suppressor oncogene, and with only a single copy of the gene, tumorigenesis is suppressed. However, a variety of neoplasms and non-neoplastic lesions are likely to develop in the absence of both copies of the gene. This NF1 oncogene concept follows the two-hit theory that is used to explain inherited retinoblastoma. The first “hit” removes one copy of the gene from the germ-cell line and therefore affects every somatic cell in the body. The affected individual is now “primed” for the second “hit” required for tumorigenesis. The loss of the only remaining protective gene probably occurs during periods of rapid cell division, for example, during embryonic and fetal growth and with the rapid growth spurt that occurs with puberty. The second hit then affects a subpopulation of somatic cells, producing the characteristic neoplastic and dysplastic manifestations. The major effects of the abnormal gene are on the nervous system (both peripheral nervous system and central nervous system) and the skin, but NF1 is a systemic disease and virtually all organ systems and tissues of the body are affected.
TABLE 5.3 Diagnostic Criteria for Neurofibromatosis Type 1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
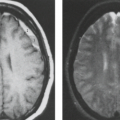
In some series, more than half of all patients with probable NF1 have abnormal neuroimaging of the brain and/or orbit (6). Both hamartomatous and neoplastic lesions occur, including optic nerve and parenchymal gliomas. Posterior fossa tumors, predominantly brainstem but with invasion of the adjacent cerebellar hemispheres, occur with a much lower incidence than supratentorial tumors (7). Malformative cerebellar anomalies have also been reported (8). Multifocal signal changes (bright foci on T2-weighted MR) in the brainstem, cerebellar white matter, dentate nucleus, basal ganglia, periventricular white matter, optic nerve, and optic pathways (9,10,11,12) (Fig. 5.1) are known as foci of abnormal signal intensity (FASI). The radiologic criteria for distinguishing these lesions as hamartomas (rather than more ominous neoplasms) are absence of significant mass effect, absence of a spreading (vasogenic) pattern of edema, and absence of contrast enhancement or hemorrhage. There have been reports of rare and transient lesion enhancement (13). MRS may aid in the differentiation of FASI and tumor in NF1, as FASI exhibit near normal N-acetylaspartate (NAA) levels (14).
Globus pallidus FASI demonstrate abnormally high signal intensity on T1-weighted images in more than half of the patients who have them and are often associated with mild mass effect. Histologic verification of FASI is rare. They most probably represent abnormal myelination, hamartomatous change, or foci of myelin vacuolization containing proteinaceous materials. Increased myelination (and increased lipid) could produce these T1-weighted hyperintensities from heterotopic Schwann cells or perhaps from ectopic melanin. Both Schwann cells and melanocytes are derived from the neural crest and while not normally present within the brain parenchyma, heterotopic Schwann cells have been seen histologically in patients with or without neurofibromatosis (9,10,15,16).
FASI peak in late childhood, and those in the basal ganglia, cerebellum, and brainstem most commonly resolve spontaneously during the teen years; they are uncommon in adults with NF1. However, lesions in the hippocampi are seen in approximately 80% of patients without any change in prevalence over time (17). Initially, globi pallidi FASI in the absence of thalamic lesions were thought to be unrelated to detectable cognitive impairment and learning disabilities (18). However, focal areas of high signal intensity have been recently found to represent imaging markers of impaired fine motor and cognitive skills when NF1 patients with FASI are compared to NF1 patients without FASI and with normal controls (19). In addition, the presence of FASI during childhood is a better predictor of adult cognitive outcome than is the presence of current signal alterations during adulthood (20). Patients with large numbers of FASI or extensive lesions at an atypical age should be followed due to the increased risk of proliferative change (21).
Optic nerve gliomas, especially those that present in childhood, are an important and often diagnostic feature of NF1 (Figs. 5.2 and 5.3). In childhood, both the sporadic ONGs
and especially those associated with NF1 are almost invariably juvenile pilocytic astrocytomas, low-grade (World Health Organization grade I) astrocytomas histologically identical to those occurring in the pediatric cerebellum as the “cerebellar juvenile pilocystic astrocytoma or cerebellar JPA.” Approximately, 2% to 5% of all brain tumors in childhood are ONGs, and about 70% of the patients with ONGs have NF1. Although almost 50% of all cases of childhood juvenile pilocytic astrocytoma (with and without NF1) present in the optic chiasm and hypothalamus, less than 3% are limited to
the intraorbital portions of the optic tract. Conversely, 5% to 40% of patients with NF1 develop ONGs, and these typically present in children younger than the age of 10 years (mean, 6 years). Later presentation is increasingly recognized in older children and young adults (7,12,22,23). Surveillance is extremely important for these young patients because up to 80% of patients with ONGs are asymptomatic at the time of initial diagnosis. The primary findings of ONG include abnormal optic nerve elongation, thickening, kinking or buckling, and abnormal enhancement (7,24). Dural ectasia of the optic nerve sheath, without a neoplasm, can mimic the enlargement and beaded configuration of an optic glioma. The presence of bilateral ONGs is considered specific for NF1 (25). There are differences between the imaging appearances of NF1 and non-NF1 patients. The orbital nerves are more likely to be involved in NF1, whereas the chiasm is the most common site of involvement in non-NF1 patients. In addition, the shape of the optic pathways is preserved, the tumor is smaller, extension beyond the optic pathway at diagnosis is uncommon, and cystic components are less common in the NF1 group (26).
and especially those associated with NF1 are almost invariably juvenile pilocytic astrocytomas, low-grade (World Health Organization grade I) astrocytomas histologically identical to those occurring in the pediatric cerebellum as the “cerebellar juvenile pilocystic astrocytoma or cerebellar JPA.” Approximately, 2% to 5% of all brain tumors in childhood are ONGs, and about 70% of the patients with ONGs have NF1. Although almost 50% of all cases of childhood juvenile pilocytic astrocytoma (with and without NF1) present in the optic chiasm and hypothalamus, less than 3% are limited to
the intraorbital portions of the optic tract. Conversely, 5% to 40% of patients with NF1 develop ONGs, and these typically present in children younger than the age of 10 years (mean, 6 years). Later presentation is increasingly recognized in older children and young adults (7,12,22,23). Surveillance is extremely important for these young patients because up to 80% of patients with ONGs are asymptomatic at the time of initial diagnosis. The primary findings of ONG include abnormal optic nerve elongation, thickening, kinking or buckling, and abnormal enhancement (7,24). Dural ectasia of the optic nerve sheath, without a neoplasm, can mimic the enlargement and beaded configuration of an optic glioma. The presence of bilateral ONGs is considered specific for NF1 (25). There are differences between the imaging appearances of NF1 and non-NF1 patients. The orbital nerves are more likely to be involved in NF1, whereas the chiasm is the most common site of involvement in non-NF1 patients. In addition, the shape of the optic pathways is preserved, the tumor is smaller, extension beyond the optic pathway at diagnosis is uncommon, and cystic components are less common in the NF1 group (26).
 FIGURE 5.1 Neurofibromatosis type 1 foci of abnormal signal intensity (FASI). A: FASI demonstrate rings of increased signal intensity (arrow) on unenhanced T1W axial image. B: FLAIR coronal image demonstrates multiple rounded foci of abnormal signal intensity (FASI) within globi pallidi (arrow), but only subtle hippocampal lesions in a child of 5 years of age. C: FLAIR axial image demonstrates increased signal FASI in the hippocampi (arrows) in the same child, now 16 years of age. The globi pallidi FASI have resolved. D: FASI are identified within the pons, right brachium pontis (arrow), and both dentate nuclei in a third child. |
 FIGURE 5.2 Neurofibromatosis type 1, visual pathway glioma. A: The intraorbital optic nerves are enlarged and tortuous (arrow) on coronal T2W image. In addition, abnormal signal of the medial frontal white matter is present. B: Enhancing chiasmatic involvement (arrow) is present on coronal enhanced fat-saturated T1W image. C, D: There is extension along the entire prechiasmatic and orbital optic nerve seen on enhanced T1W views, axial and obliqued along the plane of the optic nerve. The left orbital nerve is buckled (arrows). |
 FIGURE 5.3 Neurofibromatosis type 1, visual pathway glioma. A: FLAIR axial image demonstrate abnormal signal of the chiasm and of the retrochiasmatic optic tracts (arrow). B, C: There is extensive, bilateral signal increase and mild mass effect within the visual tracts (arrows). There is also involvement of the internal capsules, the hypothalamus, and the columns of the fornices. D: Coronal T2W image confirms involvement of the visual tract (arrow) above the choroidal fissures. There is asymmetric hippocampal involvement by FASI below the choroidal fissure. |
Serial studies demonstrate unpredictable behavior of ONG in NF1 patients, with stable or progressive growth, and even the regression of untreated ONG in patients with NF1, continuing the controversy about the management of these patients (22,25,26,27). As ONG behaves in an indolent fashion, more like a hamartoma than a true neoplasm, conservative management has previously been recommended. Treatment is currently offered with consideration of the child’s age, the size of the tumor, duration, progression or severity of symptoms and visual status (28,29,30), whereas other authors recommend a biopsy for confirmation, surgical resection, and radiation treatment (31,32,33). The use of radiation, either alone or as an adjunct to surgery, is also questioned. Irradiation of tissues that are prone to tumorigenesis (such as in patients with inherited retinoblastoma and in the phakomatoses) is relatively contraindicated because it may stimulate both the development of new neoplasms and vascular injury. Chemotherapeutic agents are increasingly being used as the first line of treatment in progressive ONG (22,32).
 FIGURE 5.4 Neurofibromatosis type 1, sphenoid dysplasia. A: CT 3D skin surface reconstruction in a 2 year old demonstrates periorbital soft-tissue swelling. B: CT 3D calvarial reconstruction reveals marked enlargement of the superior orbital fissure (arrow). The ipsilateral inferior orbital rim is depressed. C: Coronal T2-weighted image at 4 years of age shows further depression of the inferior orbital rim (arrow) by the extensive orbital plexiform neurofibroma. D: Axial fat-saturated enhanced T1W image reveals enlargement of the left middle cranial fossa, marked thinning of the sphenoid wing (white arrow) subjacent to the enhancing plexiform neurofibroma. Thinning of the lateral orbital rim (black arrow) by the plexiform neurofibroma in the temporal fossa. |
In up to one-third of patients with NF1, there are cutaneous or subcutaneous neurofibromas affecting the intraorbital and facial branches of the cranial nerves (III to VI) and/or diffuse plexiform neurofibroma of the face and eyelids, and in 5% to 10% of patients, there is proptosis of the globe because of dehiscence and dysplasia of the sphenoid bone (Fig. 5.4)
(23,24,33,34). Sphenoid dysplasia is one of the “distinctive bone lesions” that can be used as a diagnostic feature for the diagnosis of NF1 using the NIH criteria (1). This feature develops over time likely due to remodeling effect from adjacent plexiform neurofibroma (35).
(23,24,33,34). Sphenoid dysplasia is one of the “distinctive bone lesions” that can be used as a diagnostic feature for the diagnosis of NF1 using the NIH criteria (1). This feature develops over time likely due to remodeling effect from adjacent plexiform neurofibroma (35).
 FIGURE 5.5 Neurofibromatosis type 1, callosal lesions. A: FLAIR axial image demonstrates a focal area of increased signal (arrow) at 10 years of age. Focal enhancement (not shown) was present. Serial imaging showed very slow growth. B: FLAIR axial image obtained at 13 years of age reveals a large associated cyst (arrow). Biopsy revealed ganglioglioma. C: A larger splenial lesion in T2W sagittal image in another patient, 16 years old, shows a salt and pepper appearance (arrow). D: A similar speckled appearance is seen after contrast administration. This lesion has remained stable for 7 years. |
In addition to the commonly seen FASI, plexiform neurofibromas, and visual pathway gliomas, there is an increased risk for other CNS tumors. These include callosal, brainstem, and cerebellar tumors (Fig. 5.5). These tumors are predominantly pilocytic and indolent, although more malignant lesions such as anaplastic pleomorphic xanthoastrocytoma, gliosarcoma, and glioblastoma multiforme are occasionally seen (36). Radiologic progression of these lesions is quite variable, with progression, stabilization, or regression without intervention reported (37,38).
NF1 is also associated with intra- and extracranial arterial dysplasia. Intravascular aneurysms, arterial stenoses, and moyamoya occur (Figs. 5.6 and 5.7). They are reported in 2.5% of children with NF1 (39,40,41). Aneurysms have been reported in up to 9% of NF1 adult patients undergoing magnetic resonance imaging (MRI) (42). Vascular stenoses with progression to moyamoya may occur without any history of either glioma or irradiation. The frequent association of NF1 with moyamoya led Yamauchi et al. (43) to link one gene for moyamoya to chromosome 17q25. Cranial radiation for CNS tumors leads to an increased risk of moyamoya; this risk is increased threefold in patients with NF1 (44).
The presence of soft-tissue lesions, such as cutaneous, subcutaneous, and plexiform neurofibromas of the scalp, face, and neck, is variable in the pediatric NF1 population and is influenced by familial factors (Figs. 5.4, 5.8, and 5.9) (45). They typically appear late in the first decade or early in the second decade of life and frequently involve the scalp and pinnae. Children with earlier onset of skin lesions typically have larger numbers of neurofibromas covering a greater extent. Subcutaneous neurofibromas are a marker for internal plexiform neurofibromas. NF1 patients with internal plexiform neurofibromas are, in turn, considerably more likely to have malignant peripheral nerve sheath tumors than those who lack internal plexiform neurofibromas (46).
Patients with von Recklinghausen neurofibromatosis develop several different lesions affecting the spinal column and cord, including multiple nerve sheath tumors that are neurofibromas (Fig. 5.10) (rather than schwannomas, which are usually sporadic), dysplastic neural foraminal enlargement, and acquired meningoceles. The lateral thoracic meningocele is strongly suggestive of NF1 (Fig. 5.11). These lesions represent a pulsion diverticulum of the spinal subarachnoid space.
This occurs in the thorax because there are no paravertebral muscles overlying the neural foramina and because there is a larger pressure difference between the negative intrathoracic pressure and normal cerebrospinal fluid (CSF) pressure of the subarachnoid space (Table 5.3). Bony spinal abnormalities include kyphoscoliosis, which may involve a short segment and be acutely angled, progressive vertebral scalloping, and enlargement of the spinal canal (Fig. 5.11). Spinal NF, with a lack or paucity of dermal findings is considered by some authors to be a distinct phenotype of NF1. However, overlap with NF1 may occur in families (47).
This occurs in the thorax because there are no paravertebral muscles overlying the neural foramina and because there is a larger pressure difference between the negative intrathoracic pressure and normal cerebrospinal fluid (CSF) pressure of the subarachnoid space (Table 5.3). Bony spinal abnormalities include kyphoscoliosis, which may involve a short segment and be acutely angled, progressive vertebral scalloping, and enlargement of the spinal canal (Fig. 5.11). Spinal NF, with a lack or paucity of dermal findings is considered by some authors to be a distinct phenotype of NF1. However, overlap with NF1 may occur in families (47).
 FIGURE 5.6 Neurofibromatosis type 1, aneurysms. A: Coronal T2W image reveals asymmetry of the cavernous sinus (arrow) due to enlarged cavernous ICA. B: Mass-like enhancement of the right cavernous sinus is identified (arrows). C: MRA demonstrates fusiform aneurysm of the cavernous ICA (arrows). Flow disturbance is present within the ipsilateral MCA. D: Selective catheter angiography confirms fusiform aneurysm (arrows). |
 FIGURE 5.7 Neurofibromatosis type 1 (NF1), moyamoya. A: Routine surveillance T2W coronal image reveals small MCA (arrows) and absence of the flow void from the distal ICA. B: Catheter angiogram distal ICA occlusion and confirms moyamoya (arrow). C: MRA confirms lack of ACA flow on either side, small distal ICA and a pial synangiosis (arrow). |
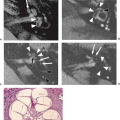
 FIGURE 5.8 Neurofibromatosis type 1 (NF1), cutaneous neurofibromas. A: A small superficial infiltrative scalp neurofibroma (arrow) is present on sagittal T1W image in this child with NF1. B: Multiple small nonenhancing nodules representing neurofibromas (arrows) are present in a different child on enhanced T1-weighted image. C: Extensive, large scalp plexiform neurofibroma demonstrates typical punctate foci, or target lesions (black arrows), on T2W sagittal image in a third child. These target lesions represent nerve fascicles surrounded by myxoid material. |
Neurofibromatosis Type 2
Formerly called bilateral acoustic neurofibromatosis, NF2 has an autosomal dominant pattern of inheritance without known racial or sexual predilection (48,49,50). It is important to note that female patients may experience exacerbation during pregnancy (51). Multiple cranial nerve schwannomas are the hallmark of this type of neurofibromatosis, and peripheral and cutaneous neurofibromas are uncommon, if they truly occur at all. The most common site for cranial nerve schwannomas, either sporadic or in NF2, is the eighth (vestibulocochlear) cranial nerve (Fig. 5.12). Schwannomas represent approximately 5% of all intracranial neoplasms; however, only 5% occur in patients with NF2. In sporadic eighth-nerve schwannomas, the tumor may be entirely limited to the vestibular division of the nerve. This localization creates the potential for hearing preservation after surgery, in addition to sparing the facial nerve function. There has been an attempt to subdivide NF2 into two types: early-onset, rapid-course (Wishart) type, named after the Scottish physician who first described the association of auditory and peripheral spinal nerve tumors, and late-onset, more benign (Gardner) type.
The NIH Consensus Committee defined a set of diagnostic criteria for NF2, as listed in Table 5.4 (50).
The NIH Consensus Committee defined a set of diagnostic criteria for NF2, as listed in Table 5.4 (50).
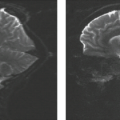
 FIGURE 5.9 Neurofibromatosis type 1, Neck. A: Coronal T2W fat-saturated image demonstrates extensive bilateral neurofibromas involving the cervical nerve roots and brachial plexus (arrows). B: In another patient, a massive prevertebral plexiform neurofibroma is present. Many typical target lesions (arrows) are seen. C: A third patient has very large bilateral vagal neurofibromas (arrows) of the neck on T2W image. D: Sagittal T2W image in the same patient demonstrates the extent of the vagal neurofibroma (arrows) from the thoracic inlet to the jugular foramen. Multiple smaller neurofibromas are seen to fill the cervical intervertebral foramina (small arrows). |
 FIGURE 5.10 Neurofibromatosis type 1, spine lesions. A: Multiple neurofibromas extend through the neural foramina (arrow), while small intradural neurofibromas (small arrows) are also seen on T1W enhanced sagittal images. B: Smaller intradural neurofibromas are present in the cauda equina of the same patient. C: Extensive rope-like plexiform neurofibromas involve the paraspinal nerves and the lumbar plexus. Typical target lesions (arrow) are present. D: Intraspinal neurofibromas (arrow) in the same child deform the thecal sac and compress the cauda equina on axial T1W enhanced image. |
 FIGURE 5.11 Neurofibromatosis type 1, dural ectasia. A: Posterior scalloping (arrow) is shown on T1W sagittal image. B: Coronal T2W image reveals outpocketing of thecal sac in areas of bony scalloping (arrows). C: Coronal T2W image in another child shows a lateral thoracic meningocele (arrow). D: Coronal T2W image in a third child reveals upper thoracic spondyloptysis (arrow). Vertebral dysplasia, dural ectasia, and plexiform neurofibromas all contribute to erosive bone changes in the spine. |
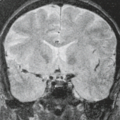
 FIGURE 5.12 Neurofibromatosis type 2, vestibular schwannoma. A: T2W axial image demonstrates typical bilateral vestibular schwannomas. The right bulges into the cerebellopontine angle and deforms the subjacent brachium pontis. The left is isointense with surrounding brain and bulges out of the porus acusticus (arrows). There is transmacular extension into the vestibule (small arrow). B: Contrast enhancement confirms the extension into the left vestibule (arrow) on T1W enhanced image. C: The intracanalicular portion fills the internal auditory canal (arrow) on the parasagittal enhanced T1 parasagittal image. D: There is marked elevation of the AICA (anterior inferior cerebellar artery) on the left (arrow) in another patient with bilateral, but asymmetric vestibular schwannomas. |
With or without NF2, the cells in some of these associated neoplasms (meningiomas and schwannomas) exhibit deletions from chromosome 22, and it was suggested in 1986 (51) and established in 1987 (52) that a loss of genetic material on this chromosome was an inherited familial trait in bilateral acoustic neurofibromatosis. Individuals who are heterozygous for the NF2 gene (at chromosome 22) are at risk for developing vestibular schwannomas, meningiomas, ependymomas, intracranial nontumoral calcification, and, less commonly, neurofibromas (49,53,54).
TABLE 5.4 Diagnostic Criteria for Neurofibromatosis Type 2 | ||
|---|---|---|
|
Evans et al. (55) reported one of the largest series of patients (150 patients) with NF2. In addition to the vestibular schwannomas, the affected patients had juvenile subcapsular lens opacities (cataracts) and a specific type of cutaneous tumor distinct from both schwannoma and neurofibroma, resembling skin tags. The diagnosed prevalence of NF2 is approximately 1 in 210,000; this translates into a calculated birth incidence for NF2 of almost 1/10 of that reported for NF1 (about 1 in 30,000 to 40,000 for NF2). Most patients (44%) present with hearing loss as the first manifestation of NF2, followed by balance problems (8%) and seizures (8%) (56). Unlike most sporadic vestibular schwannomas that merely compress the adjacent nerves, those occurring in NF2 are more likely to invade into, or even arise from, the cochlear and the facial nerves; thus, preserving hearing and facial nerve function is more difficult in patients with NF2 (56,57).
Vestibular (“acoustic”) schwannomas are the hallmark of NF2 (Figs. 5.12 and 5.13). In the absence of this congenital syndrome, vestibular schwannomas present as solitary lesions in the fifth decade; with NF2 they present as bilateral masses in the second and third decades (58). Up to 25% of cases with bilateral
vestibular schwannomas presenting over the age of 50 years are estimated to have bilateral lesions by chance rather than as a result of NF2 (59). Vestibular schwannomas almost invariably arise either within the internal auditory canal or at its orifice, the porus acusticus. Deformity and/or enlargement of the internal auditory canal are noted on routine computed tomography (CT) in more than 70% of cases (60,61). Schwannomas are histologically composed of dense areas (Antoni A tissue) and looser areas (Antoni B tissue). As they enlarge and age, schwannomas tend to become heterogeneous masses, both grossly and on imaging, because of cystic degeneration and hemorrhage (15,58,62). Schwannomas, which are the principal neoplasms of NF2, both centrally and peripherally, tend to show prominent contrast enhancement, which may be homogeneous when the tumor is small and heterogeneous when the tumor is large (63). Schwannomas are more vascular than neurofibromas, and both hemorrhage and cystic change are more common than in NF1. On nonenhanced CT, there is a tendency for both neoplasms to be hypodense compared with skeletal muscle and hypodense to isodense compared with brain (60,61,64). In addition to vestibular schwannomas, patients with NF2 develop intracranial meningiomas and schwannomas involving other cranial nerves (Fig. 5.14). Patients with NF2 may present with intracranial meningiomas that may be multiple (Figs. 5.15 and 5.16) and
can present in the lateral ventricle more commonly than sporadic meningiomas (16% vs. 5% intraventricular location) (3,7). Any meningioma presenting in childhood or young adulthood should alert the physician to be particularly suspicious for NF2 (65). The meningiomas that develop in association with NF2 are similar to sporadic tumors in their histology and radiologic appearance, differing only by their multiplicity and perhaps by a younger age at presentation. Meningiomas in the young may be identified because of symptoms related to other features of the disease or as part of family screening. Meningiomas are usually slowly growing, benign tumors that can be resected for cure. However, some researchers have suggested that the meningiomas occurring in NF2 may have a worse prognosis. The multiple meningiomas may be few and large or may appear as a myriad of small masses, producing a nodular studding along the dural surfaces. It has also been suggested that true multiple meningiomas (without accompanying schwannomas) may be an entity separate from NF2 rather than being a forme fruste of that disease, whereas others have disagreed, suggesting that most cases of multiple meningioma merely represent one end of the spectrum of NF2 (66,67).
vestibular schwannomas presenting over the age of 50 years are estimated to have bilateral lesions by chance rather than as a result of NF2 (59). Vestibular schwannomas almost invariably arise either within the internal auditory canal or at its orifice, the porus acusticus. Deformity and/or enlargement of the internal auditory canal are noted on routine computed tomography (CT) in more than 70% of cases (60,61). Schwannomas are histologically composed of dense areas (Antoni A tissue) and looser areas (Antoni B tissue). As they enlarge and age, schwannomas tend to become heterogeneous masses, both grossly and on imaging, because of cystic degeneration and hemorrhage (15,58,62). Schwannomas, which are the principal neoplasms of NF2, both centrally and peripherally, tend to show prominent contrast enhancement, which may be homogeneous when the tumor is small and heterogeneous when the tumor is large (63). Schwannomas are more vascular than neurofibromas, and both hemorrhage and cystic change are more common than in NF1. On nonenhanced CT, there is a tendency for both neoplasms to be hypodense compared with skeletal muscle and hypodense to isodense compared with brain (60,61,64). In addition to vestibular schwannomas, patients with NF2 develop intracranial meningiomas and schwannomas involving other cranial nerves (Fig. 5.14). Patients with NF2 may present with intracranial meningiomas that may be multiple (Figs. 5.15 and 5.16) and
can present in the lateral ventricle more commonly than sporadic meningiomas (16% vs. 5% intraventricular location) (3,7). Any meningioma presenting in childhood or young adulthood should alert the physician to be particularly suspicious for NF2 (65). The meningiomas that develop in association with NF2 are similar to sporadic tumors in their histology and radiologic appearance, differing only by their multiplicity and perhaps by a younger age at presentation. Meningiomas in the young may be identified because of symptoms related to other features of the disease or as part of family screening. Meningiomas are usually slowly growing, benign tumors that can be resected for cure. However, some researchers have suggested that the meningiomas occurring in NF2 may have a worse prognosis. The multiple meningiomas may be few and large or may appear as a myriad of small masses, producing a nodular studding along the dural surfaces. It has also been suggested that true multiple meningiomas (without accompanying schwannomas) may be an entity separate from NF2 rather than being a forme fruste of that disease, whereas others have disagreed, suggesting that most cases of multiple meningioma merely represent one end of the spectrum of NF2 (66,67).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


