Open spinal dysraphism: myelocele and myelomeningocele
Myelomeningocele and myelocele are the quintessential open spinal dysraphisms (open neural tube defects). In open dysraphism, neural tissue and meninges are exposed to the amniotic fluid before birth, allowing leakage of CSF and injury to the underlying neural tissue. This leads to intracranial abnormalities (the Chiari II malformation), including hindbrain herniation, hydrocephalus, loss of subarachnoid spaces, and tectal beaking. These patients have a poorer prognosis than those with closed dysraphism, with higher rates of lower extremity motor dysfunction as well as frequent bowel and bladder incontinence. The neurologic function worsens the higher and the larger the defect (8). The vast majority of these lesions are now diagnosed prenatally, with a combination of fetal ultrasound and MRI. In utero repair has recently become the standard of care and has been shown to significantly improve the hindbrain herniation, rate of ventricular shunting, and lower extremity function (9).
In the myelomeningocele, imaging findings include an expanded CSF space with the neural placode extending beyond the confines of the spinal canal through a posterior osseous dysraphic defect, the exposed neural placode and displaced meninges protruding posteriorly (Fig. 16.1).
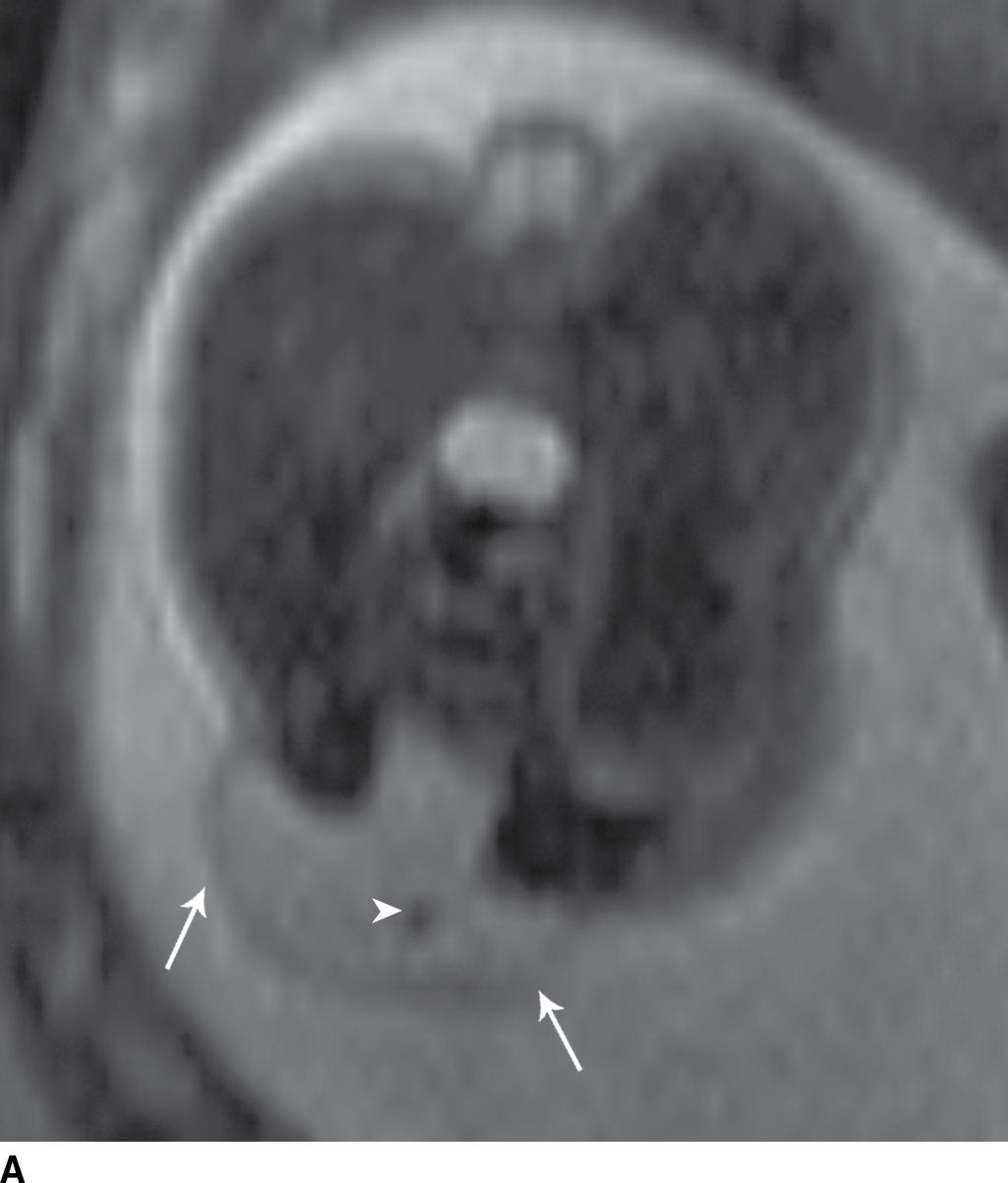
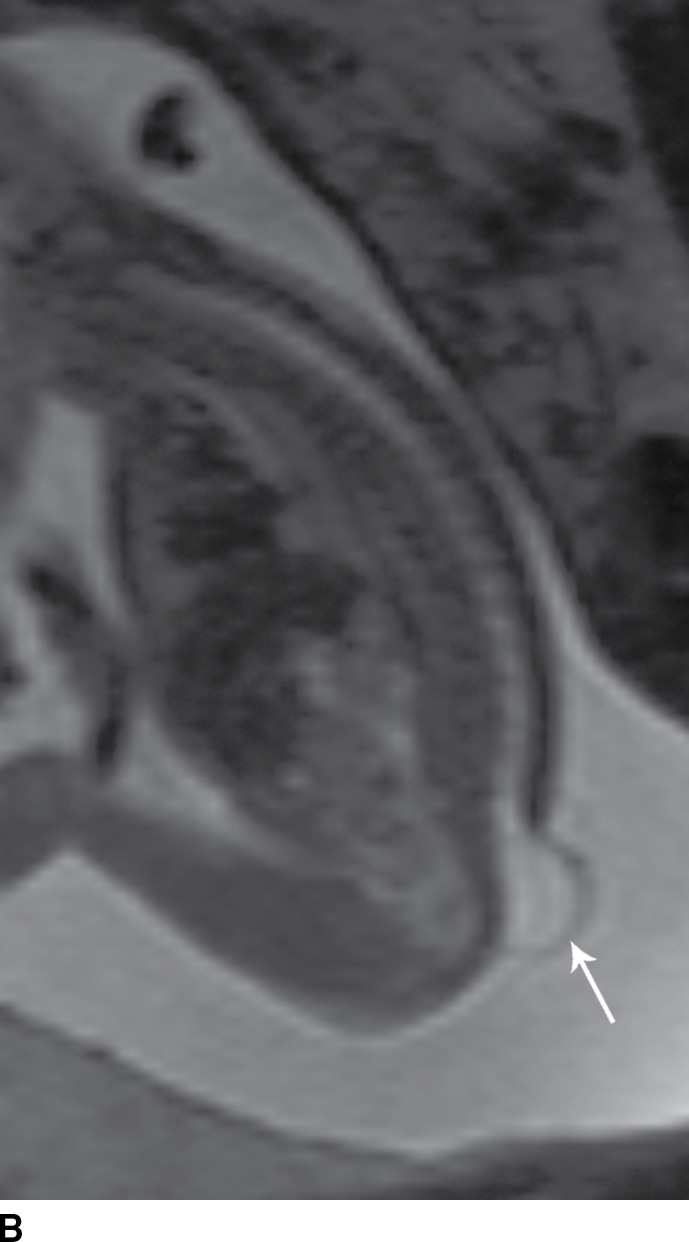
FIG. 16.1 Myelomeningocele. Fetal MRI examinations from two different patients, both performed near the end of the second trimester of pregnancy in axial (A) and sagittal (B) planes demonstrating myelomeningoceles (arrows). In the axial image, the neural placode can be visualized extending beyond the spinal canal into the myelomeningocele sac (arrowhead).
In the less commonly encountered myelocele (also referred to as myeloschisis), imaging demonstrates a neural placode that extends to but not beyond the posterior dysraphic osseous defect, there is no expansion or prominent extraspinal protrusion of the subarachnoid CSF space, and the placode typically sits within the posterior osseous defect, subjacent to the cutaneous defect (Fig. 16.2).
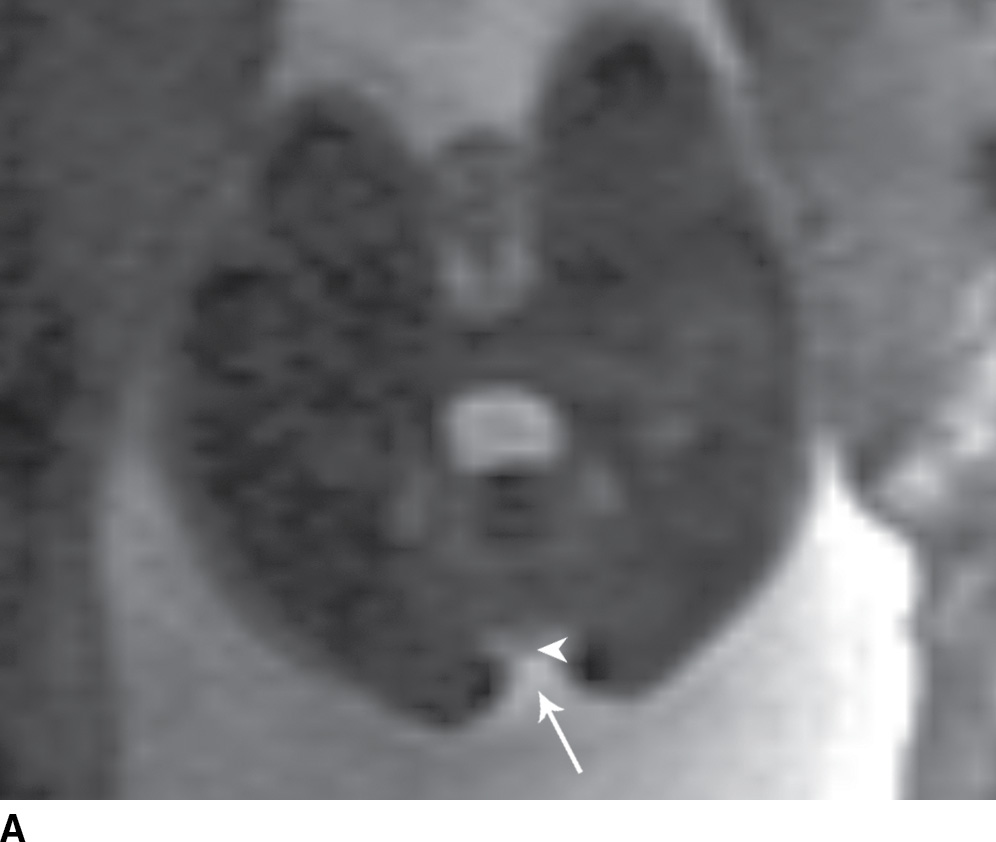
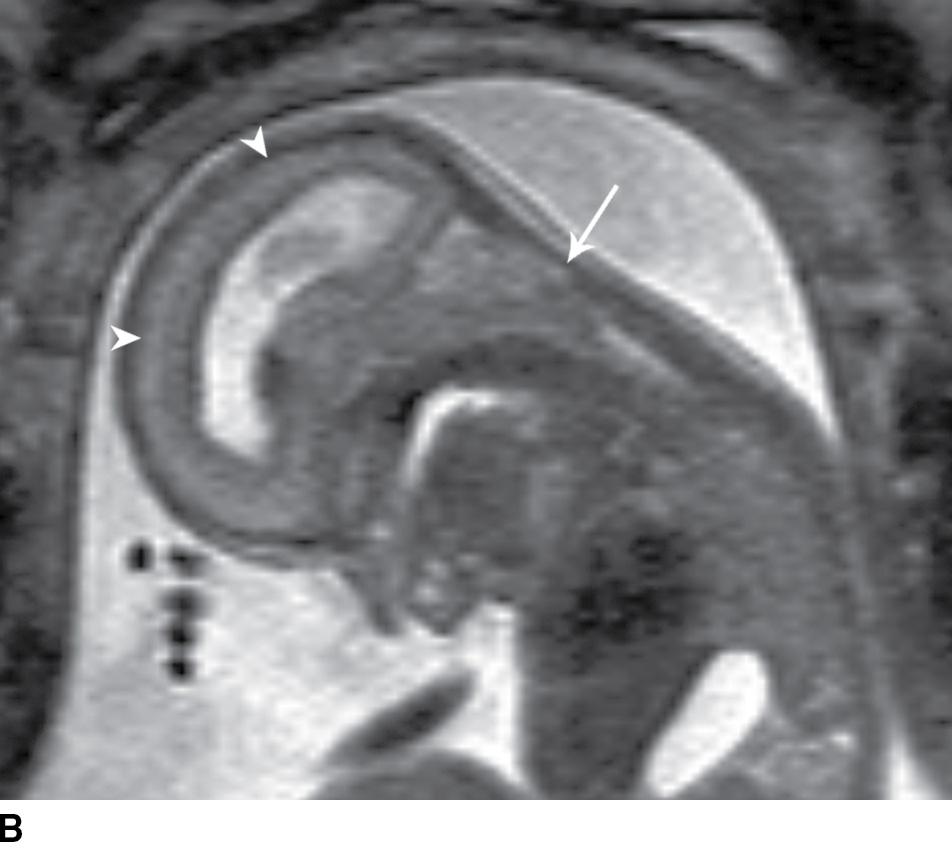
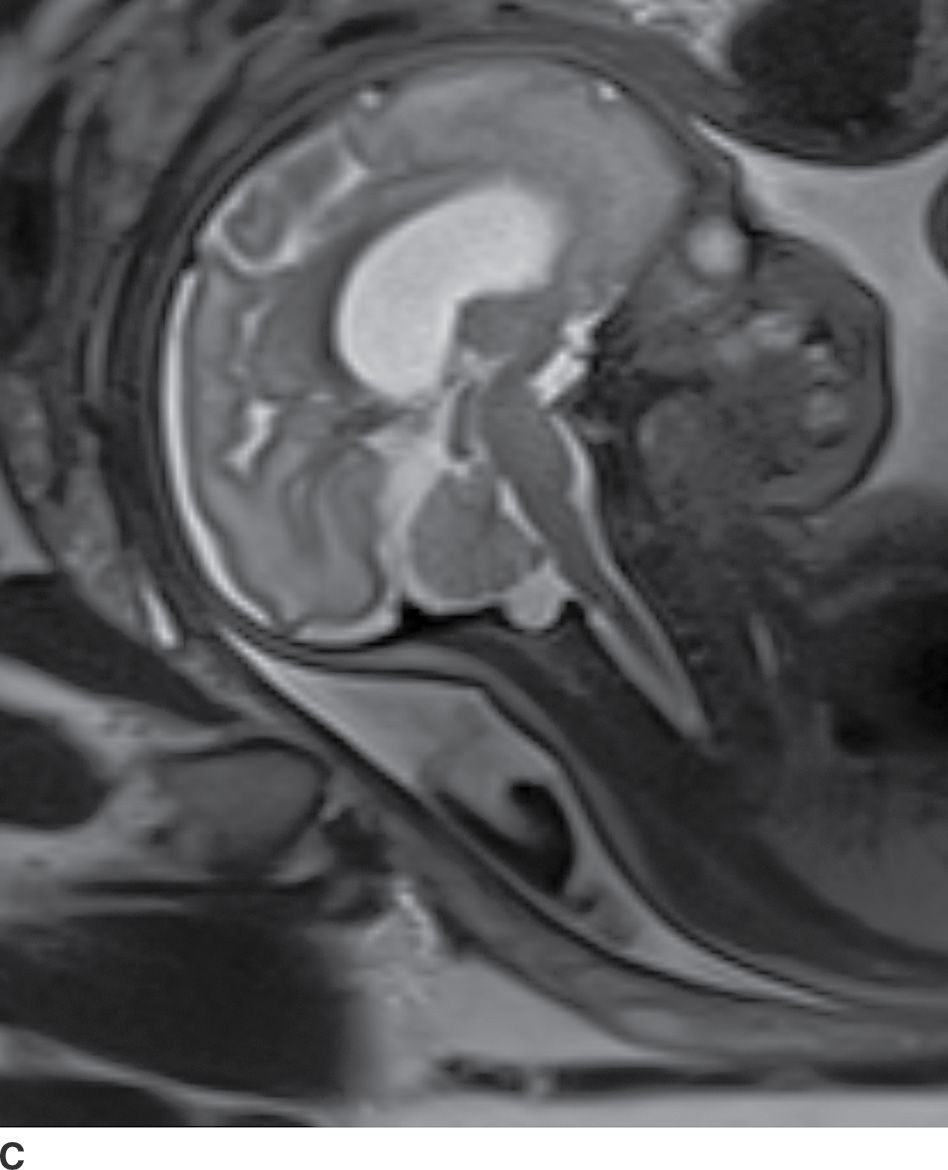
FIG. 16.2 Myelocele. A: Axial fetal MRI in the lumbosacral region at 20 weeks’ gestation demonstrates a posterior dysraphic defect (arrow) without an associated sac and without extension of neural elements (arrowhead) beyond the confines of the spinal canal. B: The sagittal MR image demonstrates the expected hindbrain herniation (arrow) and reduced CSF spaces (arrowheads) of a Chiari II malformation. C: Sagittal MR image at 32 weeks’ gestation after in utero myelocele repair at 24 weeks shows resolution of hindbrain herniation and restoration of posterior fossa CSF spaces.
Closed spinal dysraphism with a fatty subcutaneous mass: lipomyelocele and lipomyelomeningocele
Lipomyelocele and lipomyelomeningocele are closed, or skin-covered, dysraphic defects that typically present with a subcutaneous mass. Premature disjunction of neuroectoderm from cutaneous ectoderm occurs when the still incompletely tubulated neural tube separates from the adjacent cutaneous ectoderm. Mesoderm then has access through the point of disjunction to the internal surface of the unclosed neural tube and prevents tube closure. Eventually, the mesodermal elements form fat. Lipomatous hypertrophy of this subcutaneous fat can form a mass-like lipoma, which is the predominant component of the clinically evident subcutaneous mass. A posterior osseous defect is also present in the vertebrae of the affected level(s). Treatment is surgical, with many patients presenting outside of the neonatal period.
In the lipomyelocele, imaging shows a subcutaneous lipoma contacting the neural placode, which is within the confines of the spinal canal or at the level of the dysraphism (Fig. 16.3).
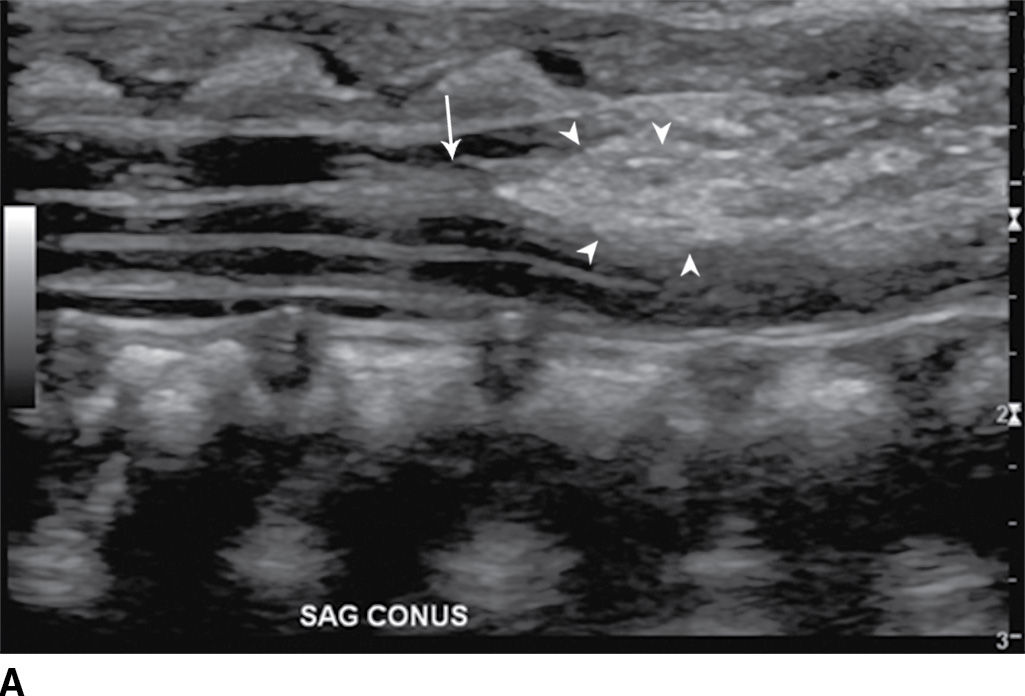
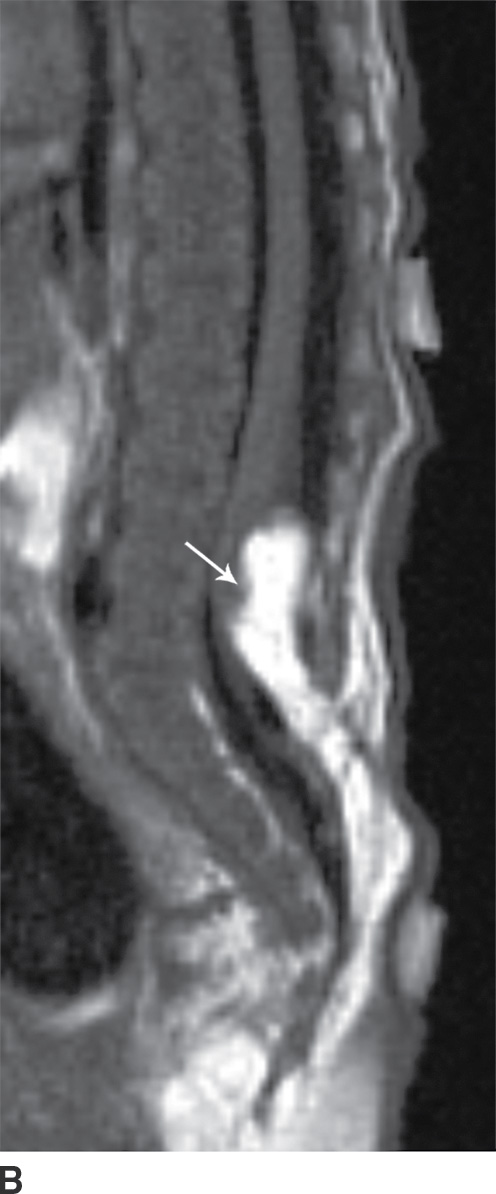
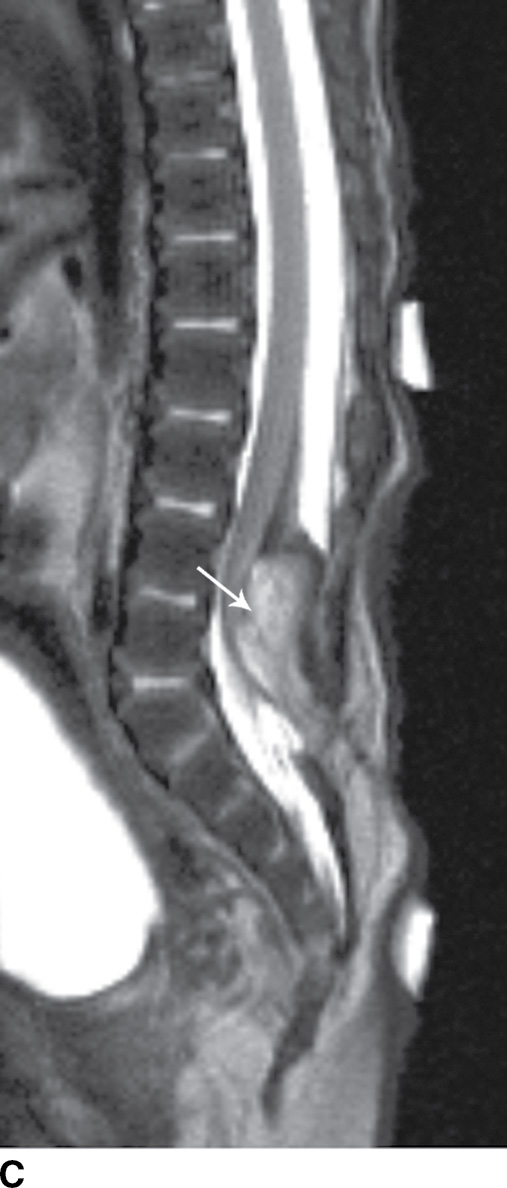
FIG. 16.3 Lipomyelocele. A 3-week-old infant who had lipoma resection and cord release procedure. Longitudinal grayscale US (A) demonstrates a low-lying conus (arrow), which abuts a globular hyperechogenic mass (arrowheads), which on sagittal T1 (B) and sagittal T2 (C) MR images has signal matching that of subcutaneous fat, consistent in appearance with a lipoma. Note that the neural placode–lipoma interface (arrows) lies within the confines of the spinal canal. Skin markers demarcate the clinically evident portion of the lesion.
In the lipomyelomeningocele, the subarachnoid CSF space is expanded, often asymmetrically, and the neural placode and meninges can be identified with ultrasound and/or MRI as protruding through the posterior osseous defect beyond the confines of the spinal canal into the subcutaneous soft tissue, with the placode–lipoma interface lying in this subcutaneous region, often in an oblique plane (Fig. 16.4).
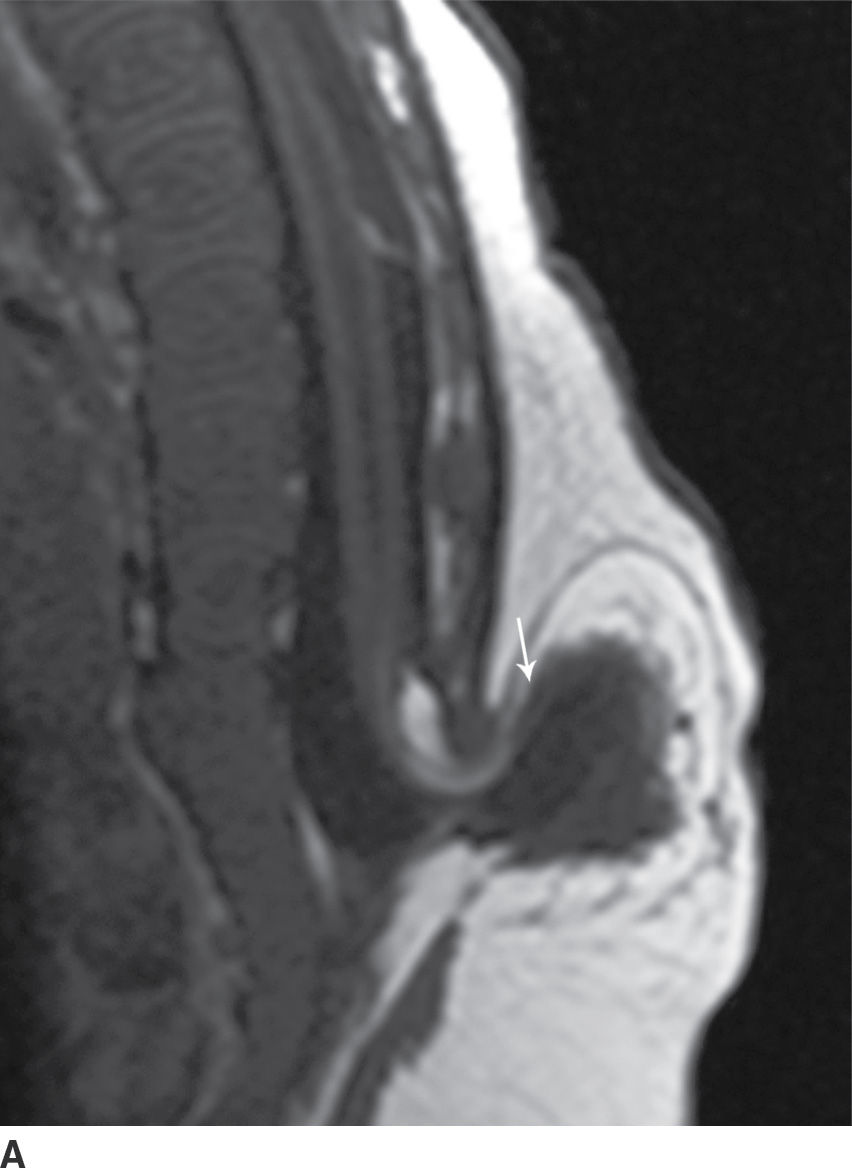
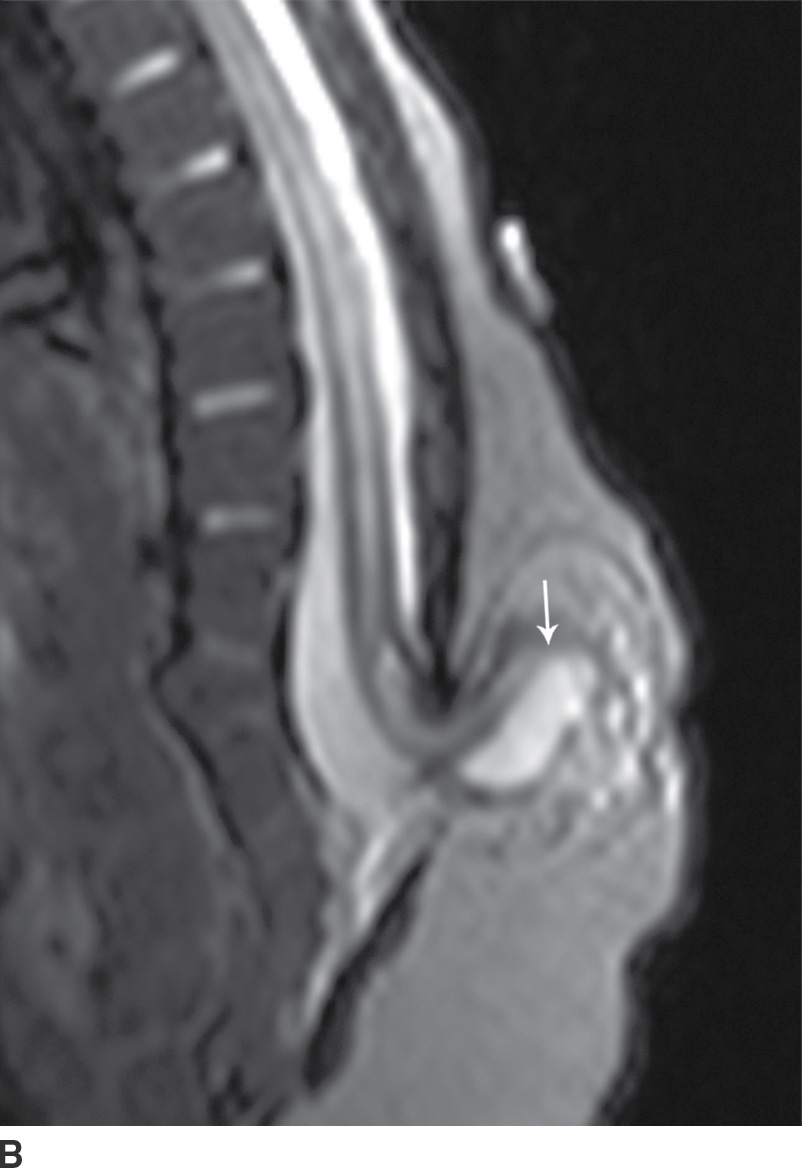
FIG. 16.4 Lipomyelomeningocele. A 2-month-old boy with a palpable back mass. Sagittal T1 (A) and T2 (B) MR images of the lower spine demonstrate a posterior dysraphic defect through which an abnormally low-lying spinal cord, meninges, and CSF herniate. The skin is intact overlying lipomatous hypertrophy of the subcutaneous fat, with a neural placode–lipoma interface (arrows) lying outside the confines of the spinal canal. A skin marker is present to facilitate vertebral counting.
Closed spinal dysraphism with a nonfatty subcutaneous mass: meningocele and myelocystocele
In the closed dysraphic state of a meningocele, there is also a subcutaneous mass, but in this case, the mass is not formed by fat. Rather, the mass is formed by herniation of the dura, arachnoid, and a CSF-filled sac beyond the normal confines of the spinal canal through an osseous defect, most commonly posteriorly. The spinal cord does not herniate into this sac in a meningocele and should be seen with US or MRI as traversing and not entering the sac. There may be associated fatty infiltration of the filum terminale, making imaging of the distal spine important (4–7,10) (Fig. 16.5).
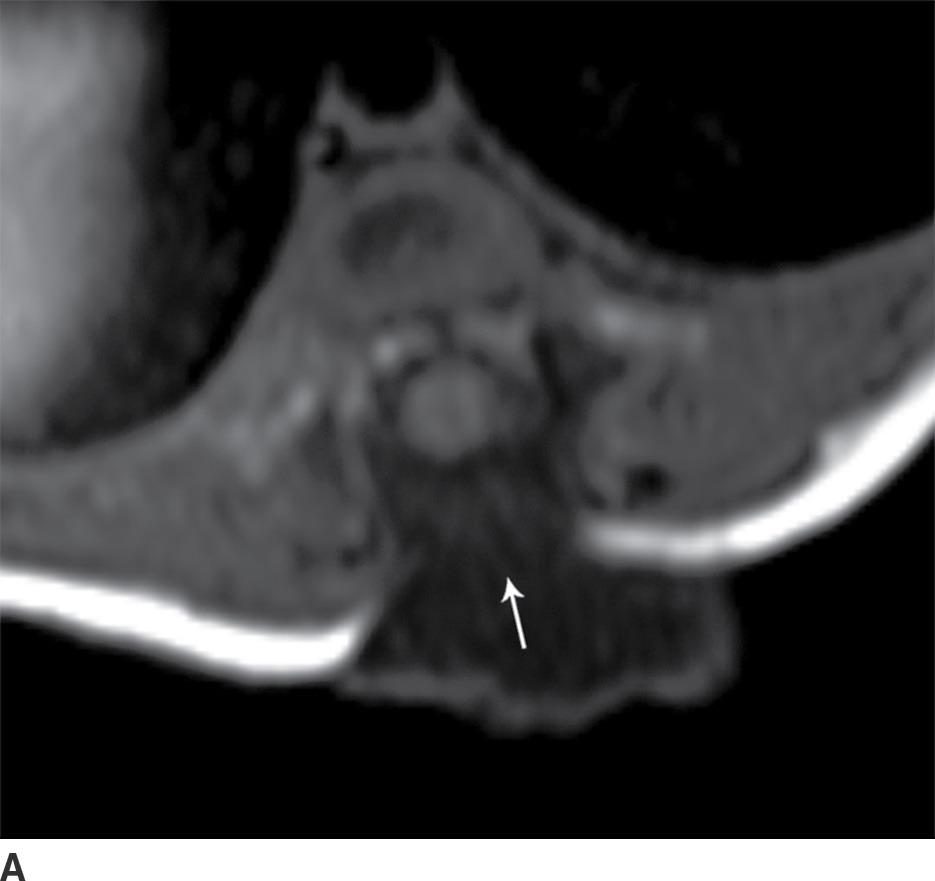
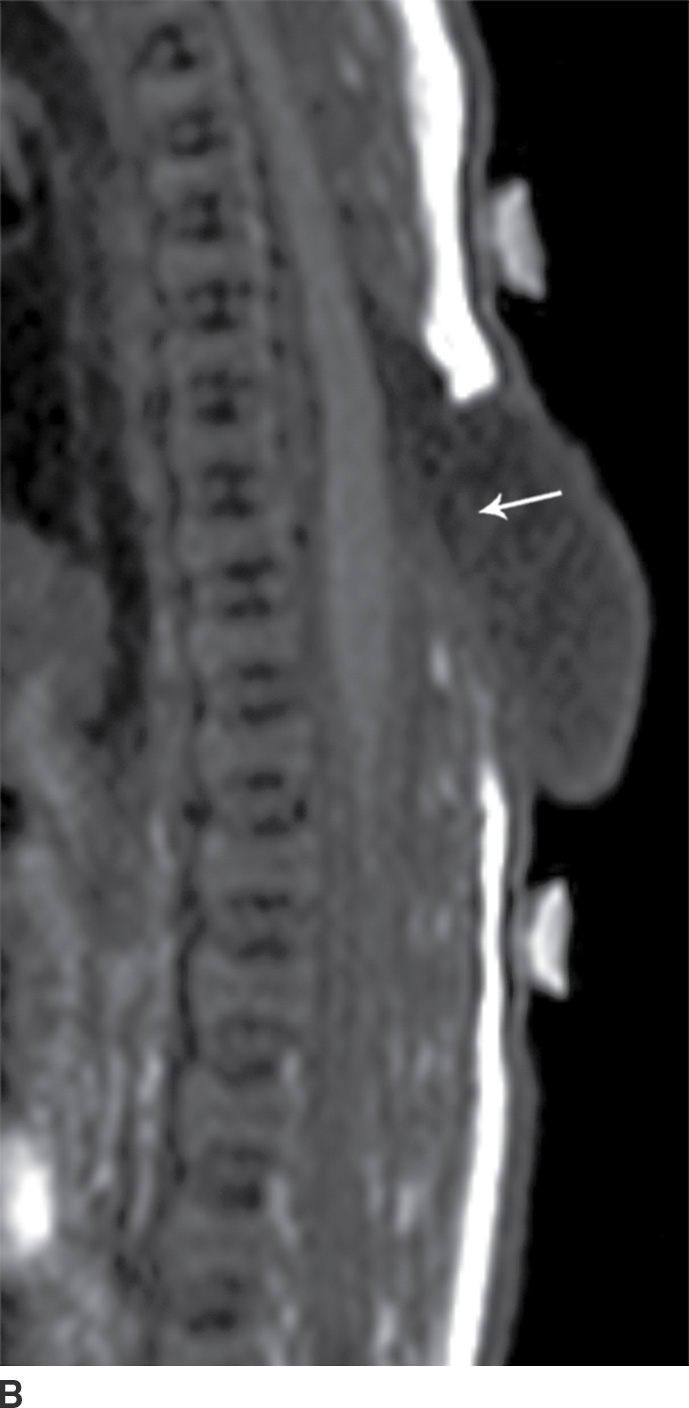
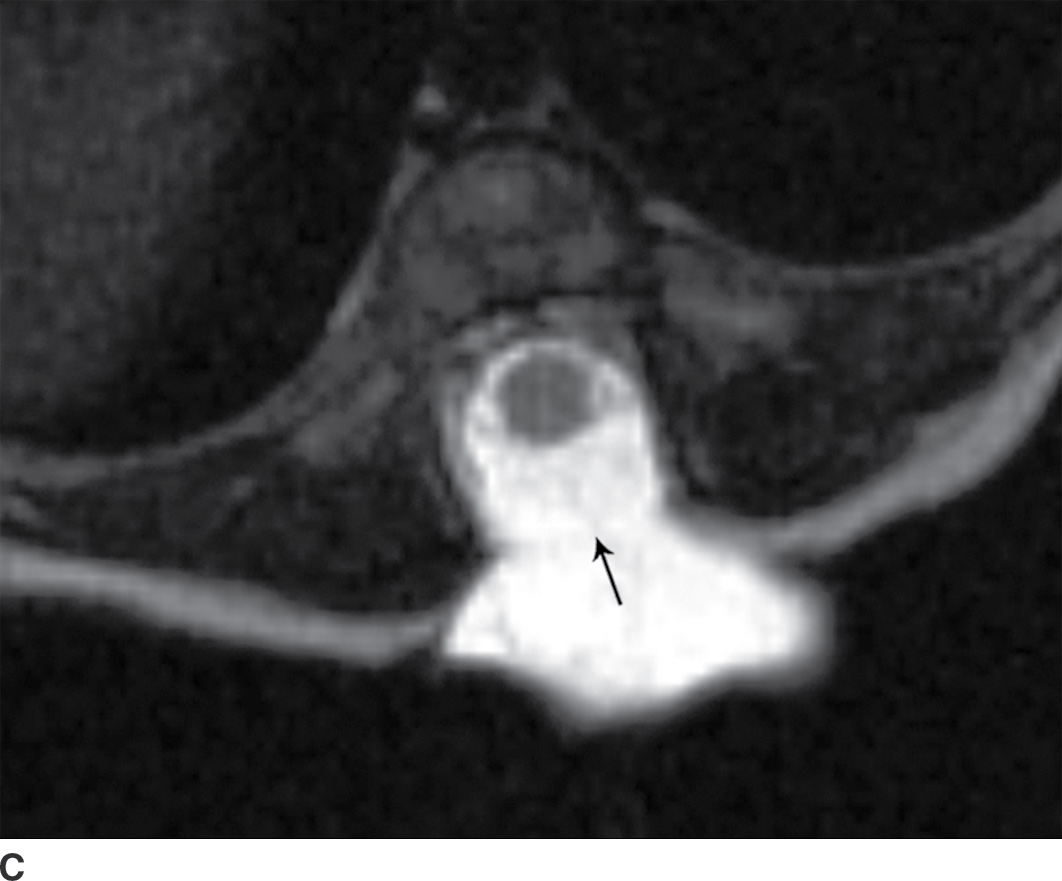
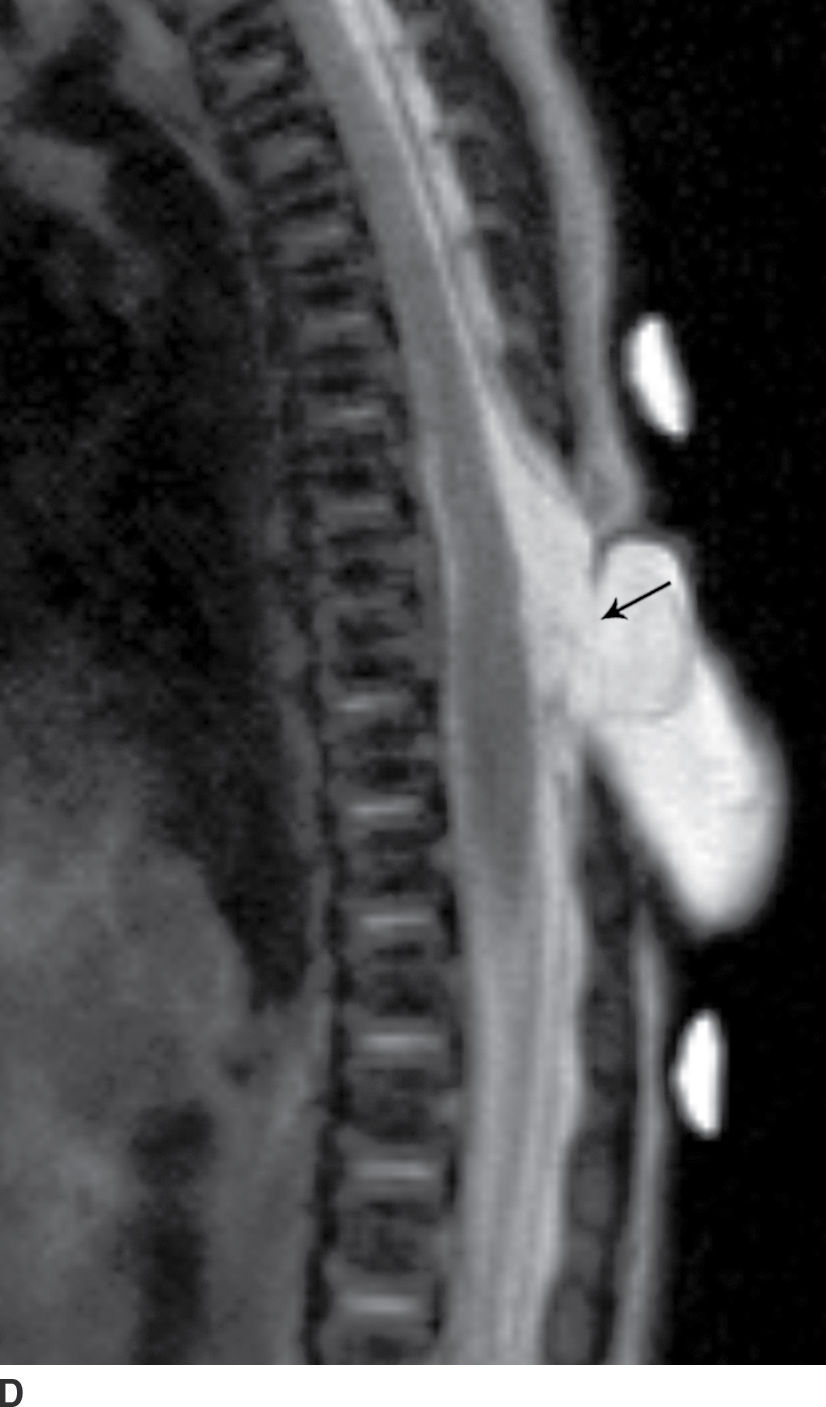
FIG. 16.5 Meningocele. A 2-day-old infant with a fluctuant back mass. Axial and sagittal T1 MR images (A,B) and axial and sagittal T2 MR images (C,D) demonstrate a dysraphic defect in the posterior elements of the spine (arrows) through which meninges and CSF herniate to form the palpable abnormality. Note that the spinal cord maintains normal course, morphology, and signal intensity. Skin markers demarcate the clinically evident portions of the lesion.
A myelocystocele is also a closed dysraphism that clinically presents with a subcutaneous mass. In this condition, however, the mass is formed by herniation of meninges and expanded CSF space as well as a portion of the spinal cord with a cyst-like dilation of the central canal of the spinal cord, often creating a “cyst within a cyst” appearance. Myelocystoceles can be terminal, involving the caudal end of the spinal cord and canal, or nonterminal, involving a more cranial aspect of the cord (4–7) (Figs. 16.6 and 16.7). Terminal myelocystoceles are frequently associated with the OEIS (omphalocele, exstrophy of the bladder, imperforate anus, spinal anomalies) complex.
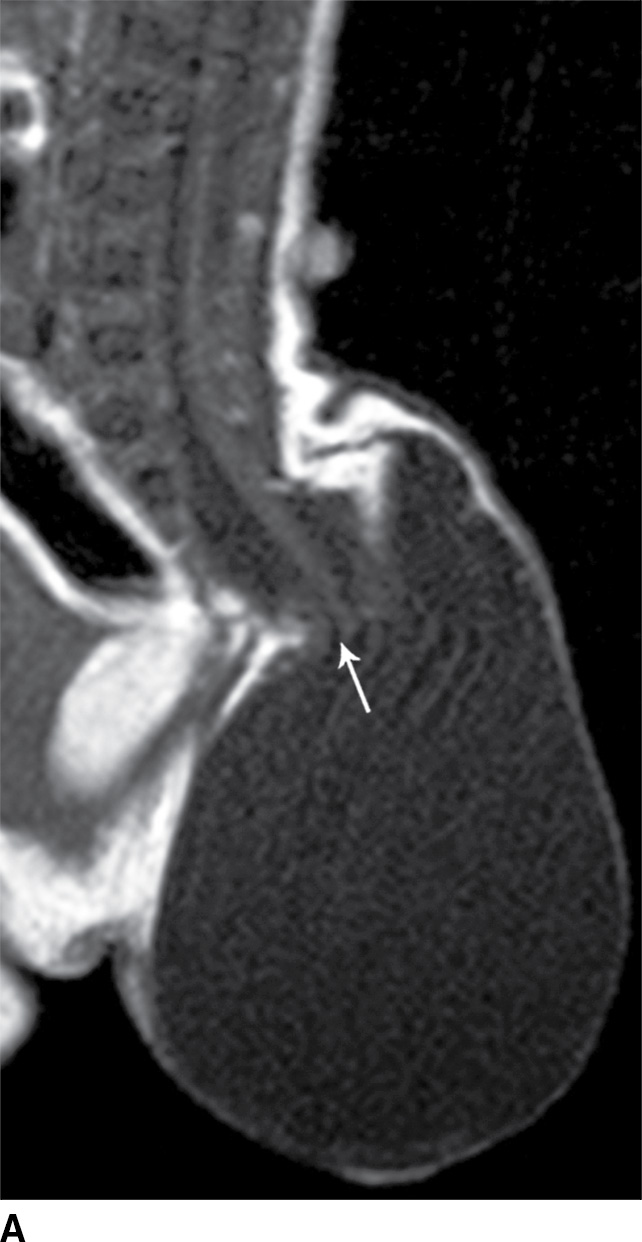
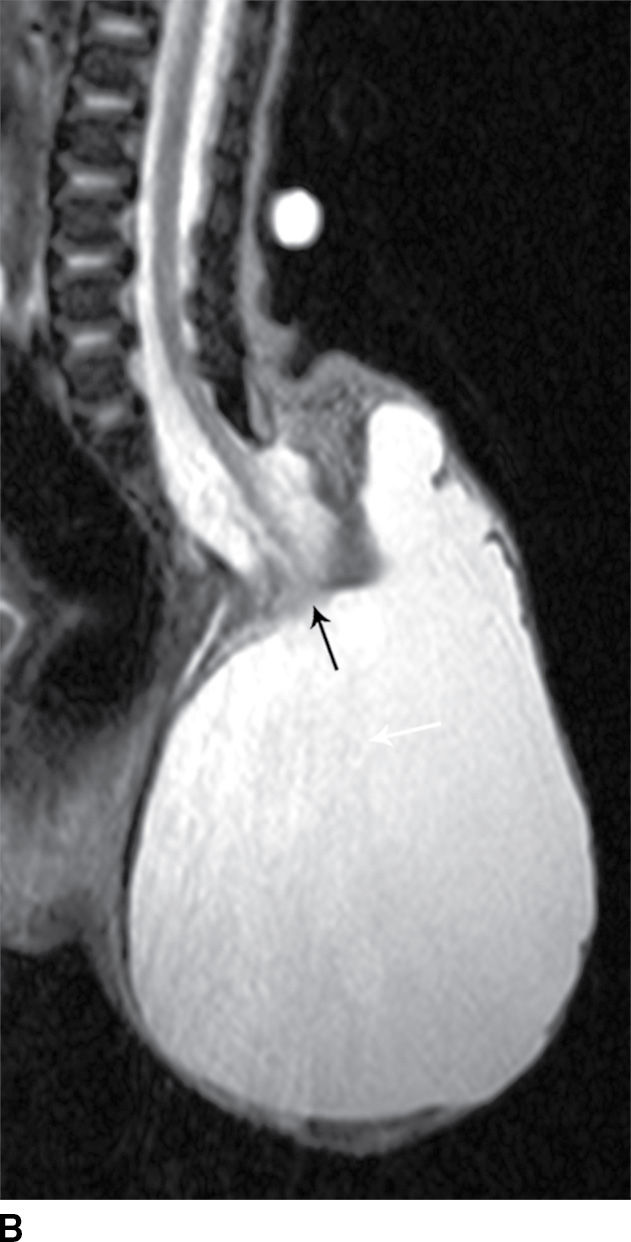
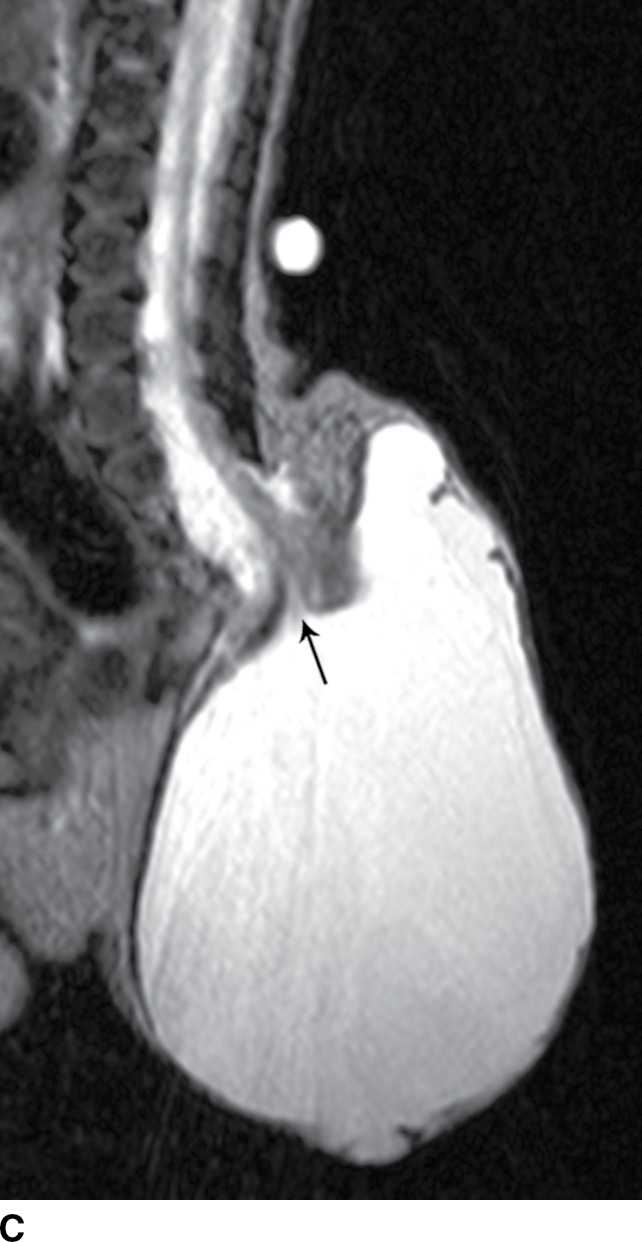
FIG. 16.6 Terminal myelocystocele. An infant with a palpable back mass. Sagittal T1 (A) and sagittal T2 (B,C) MR images demonstrate a skin-covered dysraphic defect through which herniates the abnormally low-lying cord (arrows), meninges, and CSF space. It is difficult to definitively discern that the large fluid-filled cystic space is an enlarged spinal cord central canal, but the pathologic specimen from resection demonstrated that the cystic lining had ependymal differentiation and glial tissue, confirming its identity as a myelocystocele rather than a myelomeningocele. A skin marker is present to facilitate vertebral counting.
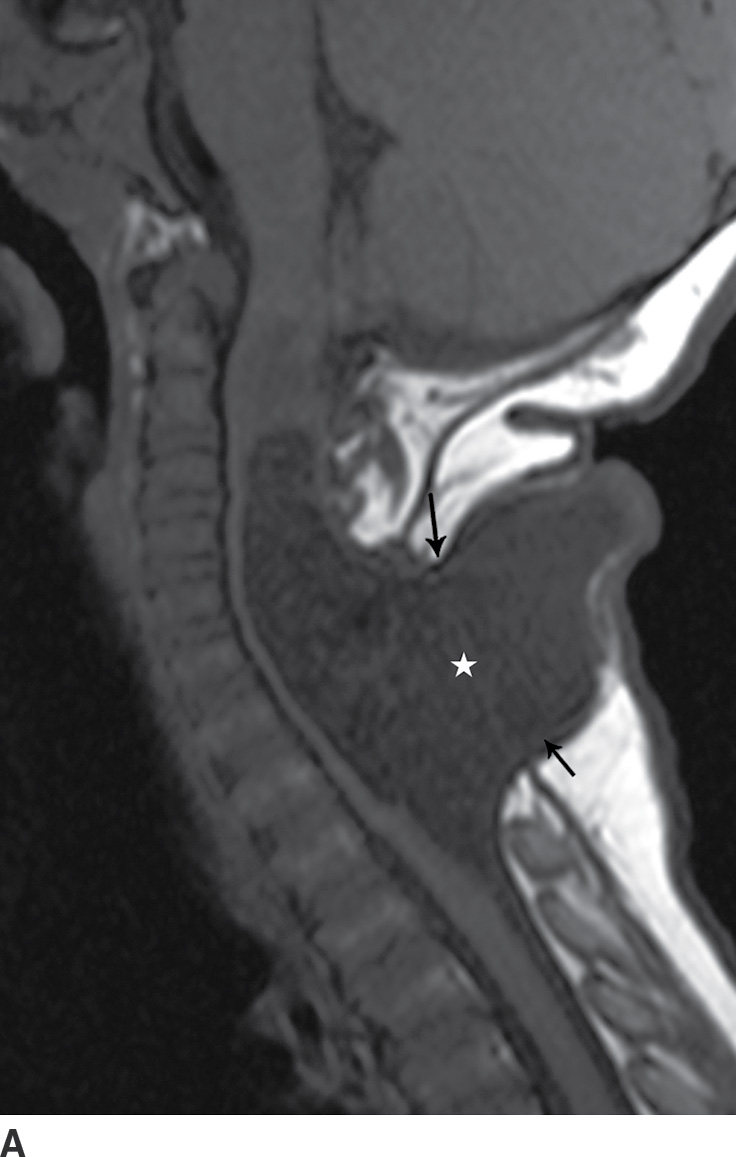
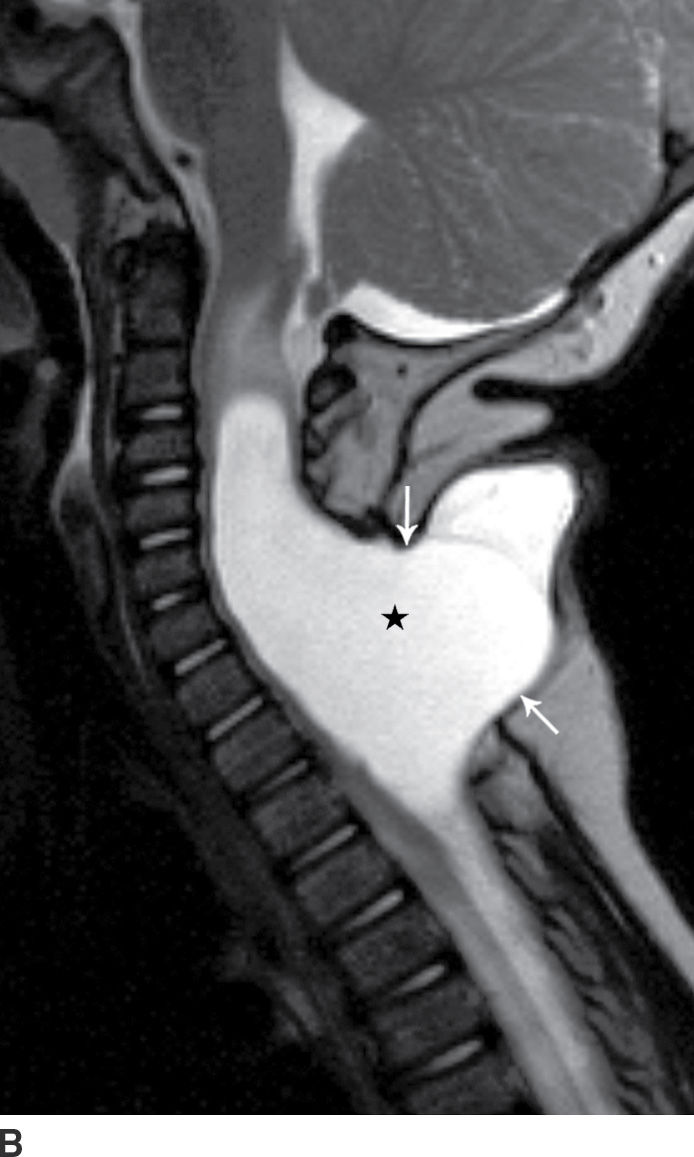
FIG. 16.7 Nonterminal myelocystocele. An 18-month-old female with a palpable posterior neck mass. Sagittal T1 (A) and T2 (B) MR images demonstrate a posterior spinal dysraphism through which herniates a portion of the spinal cord (arrows) with marked dilation of the spinal cord central canal (stars).
Entities Not Necessarily Including a Dysraphism but Often Discussed with Closed Dysraphic Disorders
Caudal regression syndrome
Caudal regression syndrome (CRS) is a rare congenital malformation characterized by varying degrees of developmental failure, with roughly 15% of affected infants having diabetic mothers (11,12). It is believed that this syndrome results from disturbances of the caudal mesoderm, including the caudal cell mass and cloaca, very early in gestation. The syndrome is composed of a spectrum of anomalies, which includes absence of caudal vertebrae and spinal cord, and may include anorectal malformations, malformed external genitalia, bladder exstrophy, renal ectopia or aplasia, neural tube defects, and lower extremity malformations (12–14). Milder forms, such as isolated sacral hypogenesis, with or without anal atresia, are more common than the more severe forms (11).
Imaging findings with CRS will vary depending upon the severity of anomalies. Patients in the more severe group have a spine that ends at S2 or above and in which there is a blunted, “wedge-shaped,” or “hatchet-shaped” spinal cord seen on sagittal imaging, with a characteristic “double bundle” shape on axial imaging. Patients in the less severely affected group, where the spine ends at S3 or below, frequently have associated stretching of the conus medullaris and a tethered filum terminale (Fig. 16.8).
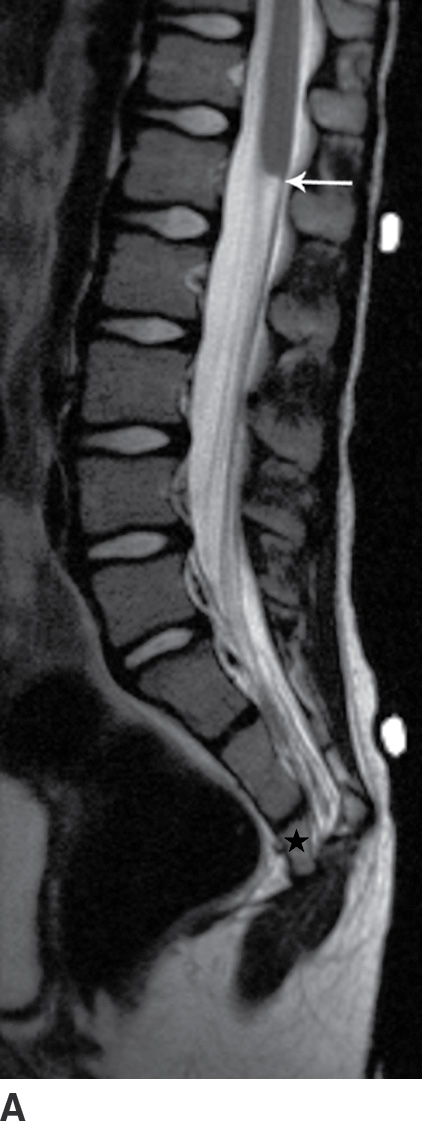
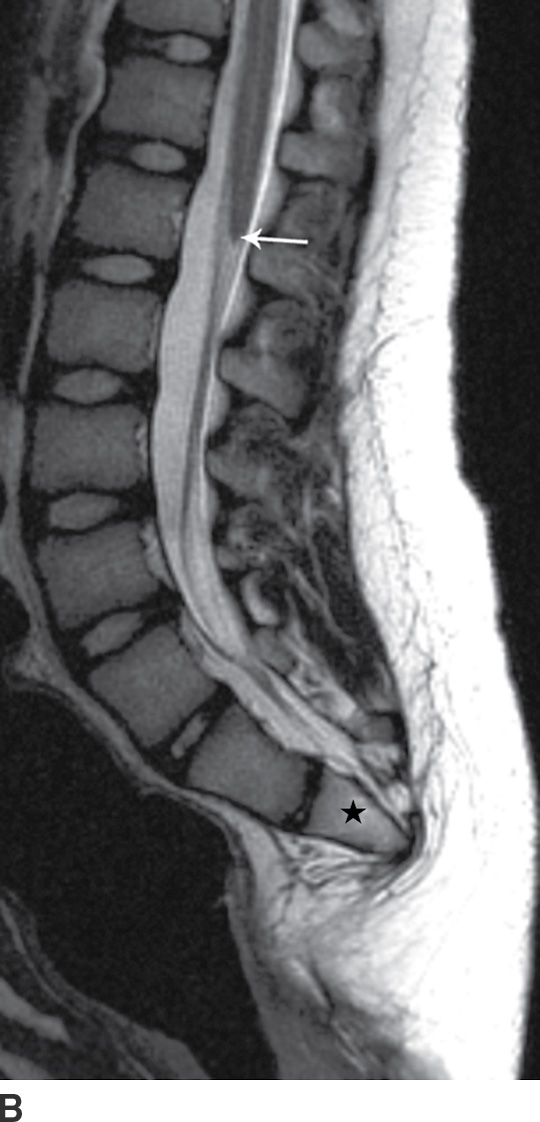
FIG. 16.8 Caudal regression syndrome. A: Sagittal T2 MRI in a 7-year-old girl with chronic bladder dysfunction demonstrates an incompletely formed sacrum (star) and a blunt ended spinal cord (arrow). Skin markers are present to facilitate vertebral counting. B: Similar findings are demonstrated in an 11-year-old boy with chronic bowel and bladder incontinence.
Segmental spinal dysgenesis
Segmental spinal dysgenesis (SSD) and CRS have been postulated to represent two facets of a spectrum of segmental malformations of the spine and spinal cord. A derangement of the caudal most mesodermal segment results in CRS, while a derangement of an intermediate segment results in SSD (15,16). Although in utero detection is possible, affected children typically are identified after birth as a result of a sharply angulated kyphotic spinal deformity, deformities of the lower limbs, hyperreflexia of the lower extremities, and/or bladder dysfunction (17–19).
Radiographs will characteristically show a moderate to severe kyphosis at the level of vertebral malformation, typically at the thoracolumbar junction with associated vertebral abnormalities (16). Three-dimensional CT reconstructions may be useful in characterizing underlying vertebral formation and segmentation anomalies (20). MRI typically demonstrates tapering of the cord to a point of marked narrowing or complete absence at the level of vertebral abnormalities. Below this point, the spinal cord may resume a normal appearance or may appear thickened (Fig. 16.9). There may be associated lipomas, dermal sinuses, or hydromyelia (16).
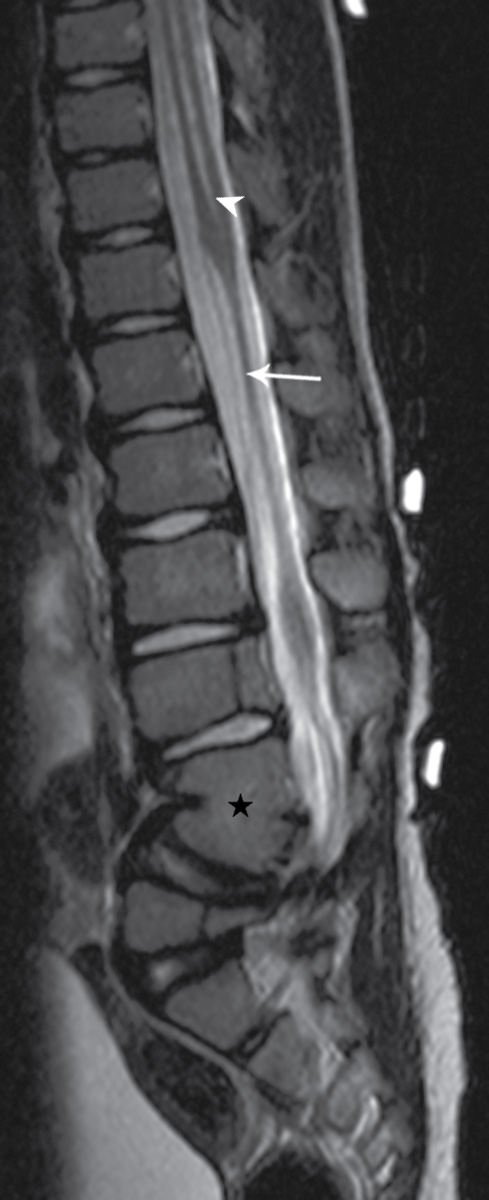
FIG. 16.9 Segmental spinal dysgenesis. Sagittal T2 MRI in a 6-year-old boy demonstrates a segment of spinal cord thinning or narrowing (arrow). Note also the central canal dilation above the narrowed portion of the spinal cord (arrowhead) and osseous anomalies below, including focal kyphosis and fusion (star). Skin markers demarcate the clinical region of concern.
Dorsal dermal sinus
The dorsal dermal sinus is an epithelial-lined tract running from the skin surface toward, and sometimes into, the spinal canal. The etiology is proposed to be a focal area of nondisjunction of cutaneous and neural ectoderm. They are most common in the lumbar region and are typically seen as tiny skin dimples or defects with associated cutaneous stigmata. A dorsal dermal sinus that communicates with the central nervous system greatly increases the risk of meningitis and/or abscess. Approximately 50% of dorsal dermal sinuses are associated with intraspinal dermoid or epidermoid inclusions.
With ultrasound or MRI, the dorsal dermal sinus will be seen as a linear, obliquely oriented tract from the skin surface through the posterior subcutaneous fat toward or frankly into the spinal canal (Fig. 16.10). Contrast-enhanced MRI is valuable in the setting of secondary infection and may help to demonstrate the dermal sinus itself, particularly when using fat suppression techniques.
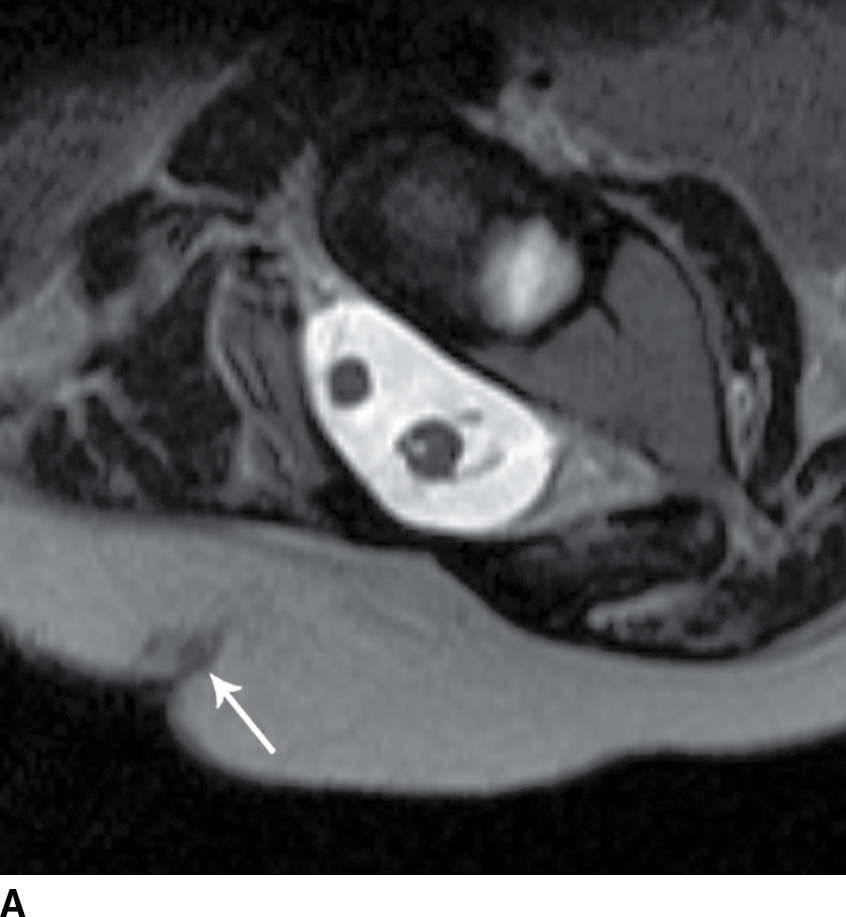
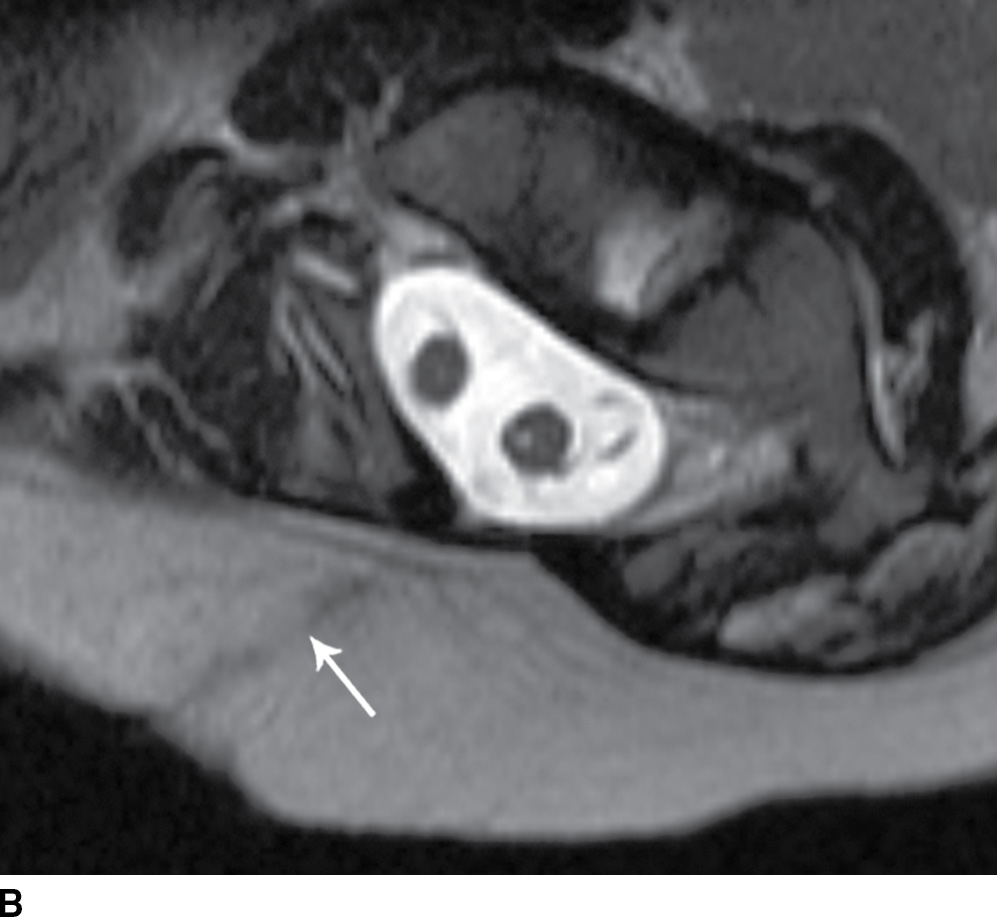
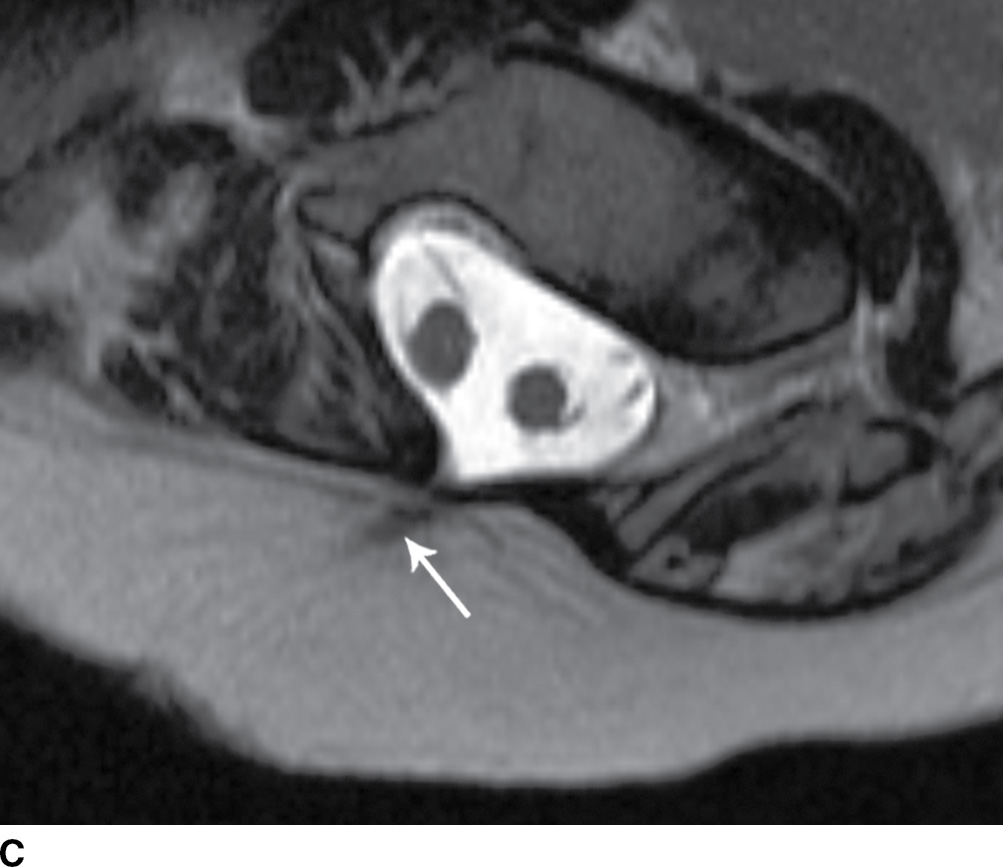
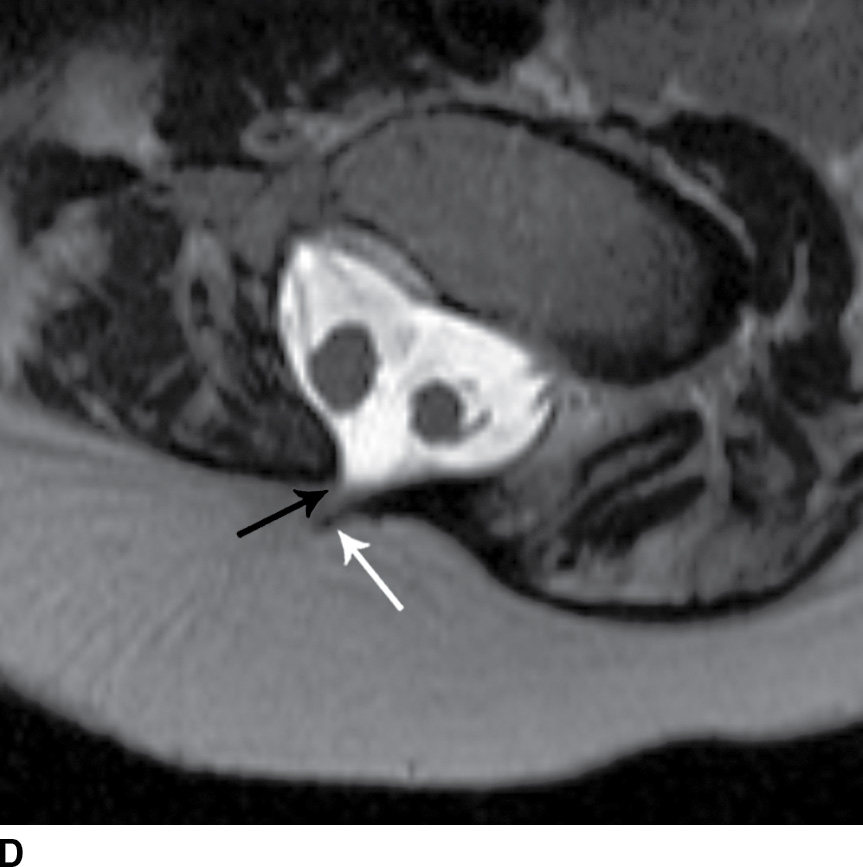
FIG. 16.10 Dorsal dermal sinus and SCM. Sequential axial T2 MR images from superior to inferior demonstrate a thin, linear, hypointense dorsal dermal sinus (white arrows) extending from the skin surface to the posterior aspect of the thecal sac through a posterior osseous defect (black arrow) in a patient with SCM. Note the tenting of the skin and thecal sac at the sites of the tract.
Split cord malformation
The term split cord malformation (SCM) encompasses diplomyelia and diastematomyelia, two entities along a spectrum of spinal cord duplication anomalies. Diplomyelia commonly refers to complete duplication of the spinal cord, in which two dorsal and two ventral nerve roots originate from each duplicated cord segment (21–25). With diastematomyelia, there is a region of sagittal division within the spinal cord with two resulting hemicord segments. Each hemicord contains a central canal, and each gives rise to one dorsal and ventral nerve root, although they may be quite asymmetrical. The division may only affect the anterior or posterior half of the cord (partial diastematomyelia or “horseshoe cord”).
SCM most likely results from splitting of the notochord during embryonic development. The notochord also influences development of the vertebral bodies, which may also explain the spinal column abnormalities, which can be present in SCM. The clinical presentation of SCM depends upon the configuration and level of the underlying malformation, but can include scoliosis, foot deformity, unilateral lower extremity muscle wasting, and low back pain (26–29). Overlying cutaneous stigmata such as hairy patches, dimples, and hemangiomas may also be present (28,29). Additional symptomatic spinal anomalies are present in the majority of patients, including spinal cord tethering, syringohydromyelia, and even open spinal dysraphism (25,27–29).
Radiographs usually demonstrate widening of the interpediculate distance and will sometimes allow visualization of a midline osseous septum in patients with SCM (28). CT is particularly helpful in detailing the anatomy of the osseous anomalies and septum. MRI is the most sensitive examination for the anatomy of the spinal cord and canal, optimally showing the two hemicords, either in a single- or dual-dural tube.
Tethered Spinal Cord
The term tethered cord syndrome is used to describe a spectrum of congenital anomalies that may result in an abnormally low termination of the conus medullaris and potentially associated neurologic, musculoskeletal, urologic, and gastrointestinal abnormalities (30). Although the term is most commonly used in reference to patients with a low-lying conus medullaris and a thickened filum terminale, many of the previously described spinal anomalies and cord abnormalities may result in a “tethered” spinal cord. These abnormalities include open spinal dysraphism, lipoma with dorsal defect, filar lipoma, dorsal dermal sinus, and SCM (30,31) (Fig. 16.11). The true incidence of tethered cord syndrome is not known, but the number of patients diagnosed with this condition is increasing due to incidental detection with more routine use of MRI and greater awareness of these entities (32).
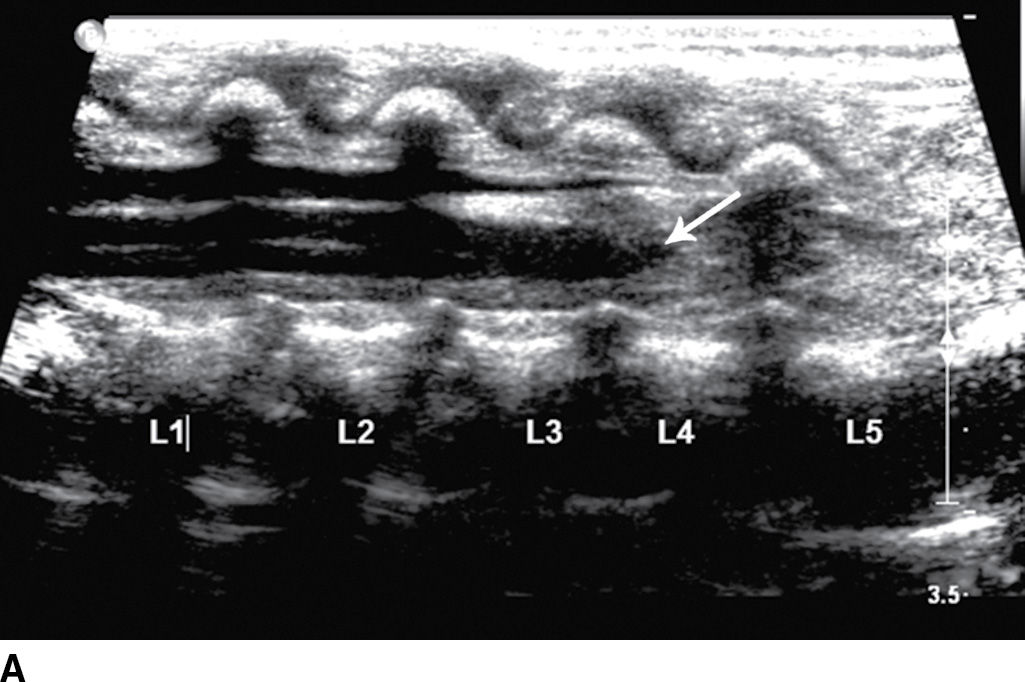
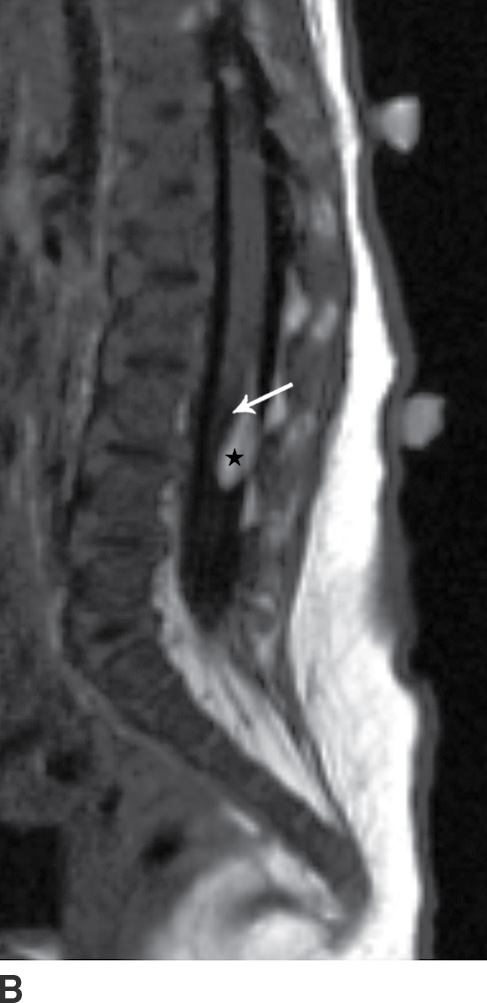
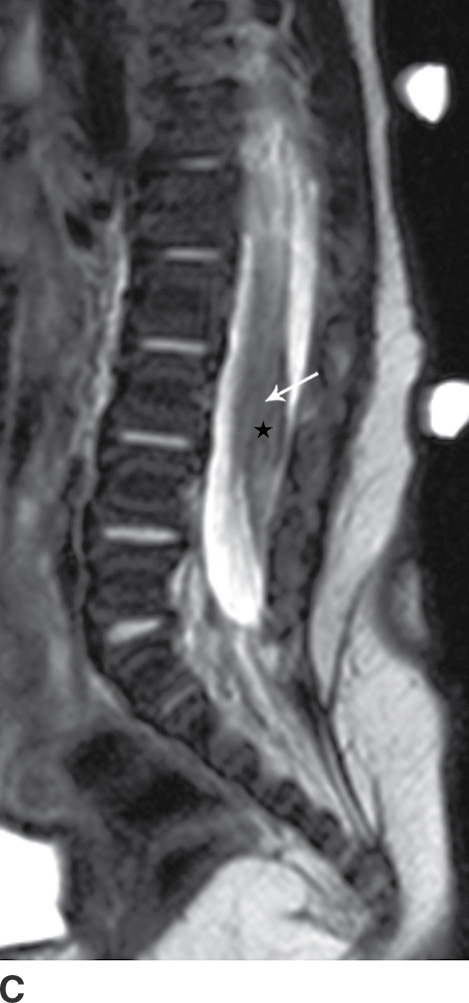
FIG. 16.11 Low-lying conus medullaris and intradural lipoma. A 2-week-old infant with a sacral dimple and skin tag. A: Longitudinal grayscale US shows a low-lying conus medullaris with its tip at the L4 level (arrow). Sagittal T1 (B) and T2 (C) MR images also demonstrate the presence of a globular T1 hyperintense focus adjacent to the low-lying conus consistent with an intradural lipoma (star). Skin markers are present to facilitate vertebral counting.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


