Fig. 1
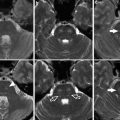
“Ivy sign” within the leptomeninges on T2 FLAIR (thin arrows) and white matter changes related to silent ischemia often seen in watershed areas (thick arrows)
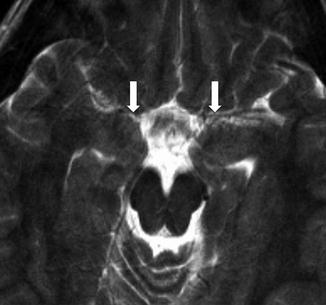
Fig. 2
An 8-year-old with headaches. Decreased visualization of normal MCA flow voids on T2 (arrows)
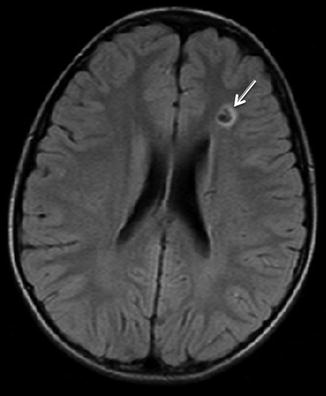
Fig. 3
Same patient as Fig. 2. Eight-year-old with headaches. T2 FLAIR image demonstrates a focus of chronic white matter ischemia in the left ACA/MCA watershed territory (arrow)
On MRA, narrowing of the distal ICAs and/or the proximal MCA/ACAs can be seen (Fig. 4). MRA may show basilar collateral formation. Digital subtraction angiography will show these findings well, demonstrating the characteristically described “puff of smoke” as the basilar collateral formation causing thin wispy densities to appear like strands of smoke (Fig. 5).
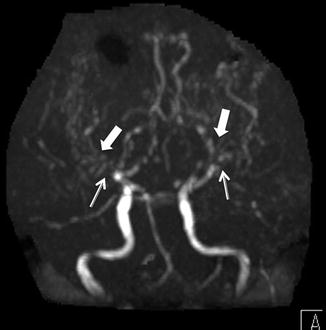
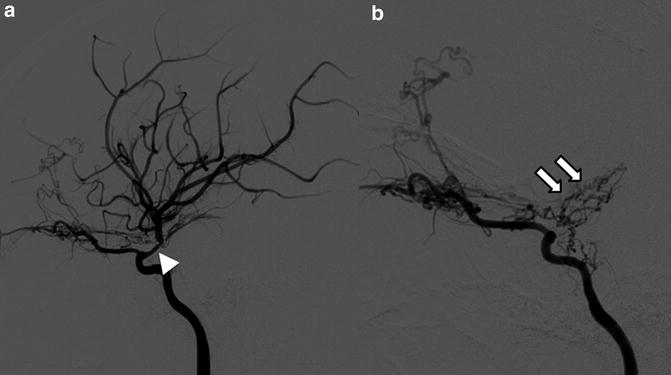
Fig. 4
Time of flight MRA, anterior projection MIP, in a 10-year-old with moyamoya demonstrates the absence of flow in MCAs (thin arrows) and multiple tiny collaterals (thick arrows)
Fig. 5
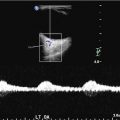
Eleven year old with new stroke found to have moyamoya. (a). At initial diagnosis, right ICA injection (lateral) projection, showing narrowing of MCA (arrowhead). (b). Follow-up 2 years later demonstrates complete occlusion of MCA with formation of basilar collaterals, aka “moyamoya vessels” (arrows)
TREATMENT: The treatment for moyamoya is a revascularization procedure. Many centers are utilizing indirect or direct techniques to create collaterals or reestablish arterial blood supply to the cerebral areas at risk for infarct. A current common technique is an indirect revascularization, encephalo-duro-arterio-synangiosis (EDAS), which can be combined with other techniques such as pial synangiosis, dural inversion, or split dural techniques in order to optimize collateral formation (Fig. 6). Some centers may include perfusion imaging techniques to determine presurgical and postsurgical appearances.
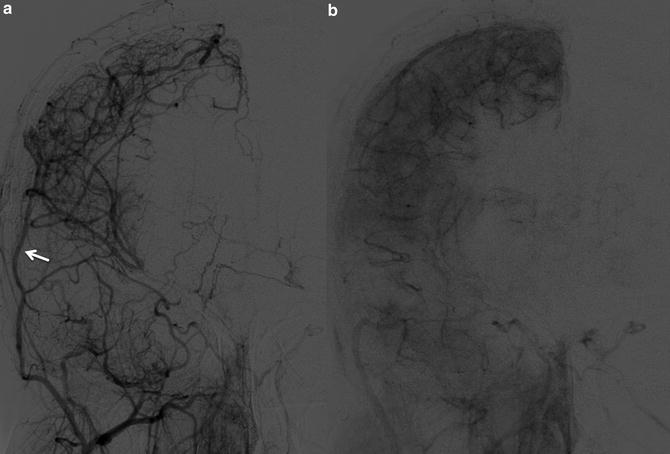
Fig. 6
Early (a) and delayed (b) images from ECA injection (AP projection). 13-year-old with moyamoya, status post revascularization with pial synangiosis using right superficial temporal artery (arrow)
PHACE Syndrome
The presence of an infantile hemangioma in the head and neck region, particularly if in a segmental distribution, should raise the clinical concern for associated intracranial and vascular abnormalities. The acronym PHACE (or PHACES) represents the spectrum of findings in this rare neurocutaneous condition.
Posterior fossa/intracranial abnormalities
Hemangioma (cervicofacial)
Arterial cerebrovascular abnormalities
Cardiac abnormalities and aortic coarctation
Eye abnormalities
PHACE association is listed in Online Mendelian Inheritance in Man (OMIM 606519) and may be termed PHACES when referring to the association with the involvement of Sternal cleft or supraumbilical raphe abnormalities.
Patients with hemangiomas of the head or neck often undergo evaluation for PHACE syndrome with imaging examinations. It is important to understand the diagnostic imaging criteria for making the clinical diagnosis. MRI of the brain with and without contrast to evaluate for structural abnormalities as well as optimized MRA of the aorta, neck and head with and without contrast are important for accurate determination of arterial abnormalities commonly seen in PHACE patients.
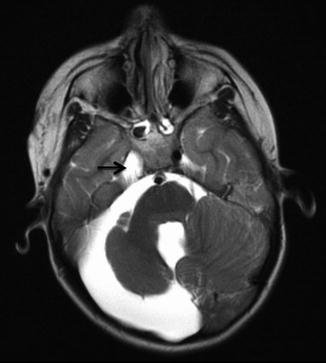
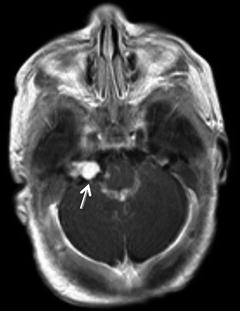
Posterior fossa/intracranial abnormalities – A major criterion for PHACE syndrome is posterior fossa abnormalities. This is most typified by cerebellar hypoplasia. This may manifest as unilateral cerebellar hemispheric hypoplasia, usually ipsilateral to the cervicofacial hemangioma, however the hypoplasia may be bilateral or involving the cerebellar vermis (Fig. 7). Intracranial hemangiomas have typical imaging appearances for infantile hemangiomas and are considered minor criteria (Fig. 8). Other structural anomalies, in particular midline and neuronal migration abnormalities, are considered minor criteria.
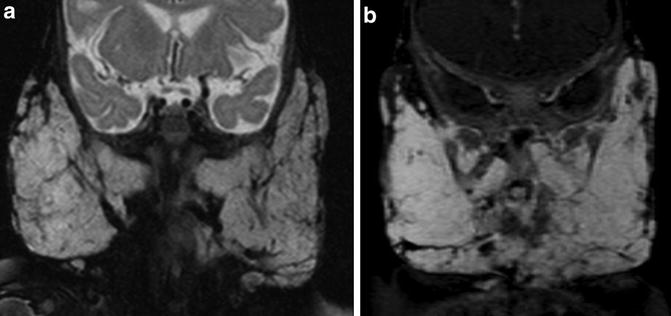
Hemangioma – The infantile hemangioma is the most common tumor in infancy and typically presents within the first few weeks of life, has a rapid proliferative phase within the first year of life, then begins spontaneous regression. As defined in a consensus statement published in 2009, PHACE syndrome requires a facial hemangioma >5 cm (plus one major or two minor criteria). Possible PHACE syndrome may have only one minor criterion or may include a chest/neck hemangioma with one major or two minor criteria or may have no hemangioma if two major criteria are present. Imaging features of hemangioma include significant T2 hyperintensity, presence of flow voids, and early arterial enhancement with persistent blood pool enhancement (Fig. 9). During the proliferative phase, lesions are homogeneous in enhancement and well defined. The presence of an infantile cutaneous lesion with similar intense enhancement but with infiltrative or aggressive imaging features should question the diagnosis of hemangioma and raise concern for a kaposiform hemangioendothelioma.
Arterial cerebrovascular abnormalities – Craniocervical arterial abnormalities are major criteria in PHACE syndrome. Common abnormalities include congenital absence, excessive tortuosity, looping, kinking, dolichoectasia, aneurysm or stenosis of a portion of the carotid, vertebrobasilar, or intracerebral vasculature (Fig. 10). Moyamoya vasculopathy has also been reported. Persistent embryonic arterial vasculature is also common, with persistent trigeminal artery being a major criterion for PHACE. While arterial strokes have been reported, they are rare but reflect a significant possible sequela of the syndrome.
Cardiac abnormalities and aortic coarctation – Aortic arch anomalies are a major criterion, with dysplasia and coarctation being the classic appearance (Fig. 11). Aberrant subclavian origin and aortic aneurysm are also considered major criteria. Ventricular septal defects and presence of a right-sided aortic arch are minor criteria.
Eye abnormalities – Posterior segment abnormalities are major criteria, specifically persistent hyperplastic primary vitreous (PHPV), retinal vascular anomalies, morning glory disc anomaly, optic nerve hypoplasia, peripapillary staphyloma, and colobomas. Anterior segment abnormalities are minor criteria, specifically cataract, anterior coloboma, sclerocornea, and microphthalmia.
Fig. 7
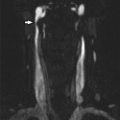
Axial T2-weighted image in child with right facial hemangioma demonstrates ipsilateral cerebellar hypoplasia, enlarged posterior fossa, and absence of ipsilateral internal carotid artery (arrow)
Fig. 8
Three-month-old infant with bilateral facial hemangiomas with right posterior fossa hemangiomas, largest at the right porus acousticus (arrow)
Fig. 9
Six-month-old girl with bilateral facial masses. (a). Coronal T2 fat sat image demonstrates hyperintense bilateral parotid masses with internal flow voids. (b). Intense homogeneous enhancement of the masses is seen on coronal T1 fat saturated post contrast image
Fig. 10
Axial image from noncontrast time of flight MRA shows significant tortuosity of the left ICA
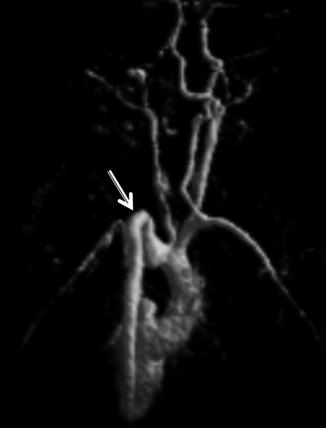
Fig. 11
3D MRA reconstruction (posterior view) demonstrating aortic dysplasia, kinking, and coarctation in an infant with PHACES
TREATMENT: Multiple therapeutic options can be utilized for the management of infantile hemangiomas, including steroids, surgical resection, laser treatments, and most recently the use of beta blockers, in particular propranolol. Hemangioma therapy may be related to size and location of the mass. Additional treatments are related to the individual patient’s manifestations of PHACE syndrome, particularly cardiovascular abnormalities. While the cerebrovascular abnormalities are well-defined in the literature, the long-term course and necessity for treatment and management of intracranial arteriopathies are being evaluated and are typically assessed by individual case review. Recommendations for routine follow-up imaging relate to severity of arteriopathy and concern for progression of disease and potential need for treatment.
AVM/AVF Syndromes
In reference to the terminology of vascular malformations, reference to ISSVA classification is recommended. In general, arteriovenous malformations are considered congenital disorders, with underlying etiology of the malformation due to abnormal developmental communications of arteries and veins. Classification and discussion of AVMs and AVFs are covered more extensively in Chapter “Brain Arteriovenous Malformations”.
Many children may have CNS abnormalities in association with their syndrome/disease or as reportable variant anomalies. The syndromes below are highlighted because of their primary neurovascular manifestation.
Heriditary Hemorrhagic Telangiectasia
Heriditary hemorrhagic telangiectasia (HHT), also known as Rendu-Osler-Weber or Osler-Weber-Rendu disease, is an autosomal dominant condition that manifests as mucocutaneous telangiectasias and visceral arteriovenous malformations. Central nervous system abnormalities in HHT are less common than involvement of the body. Arteriovenous malformations and cavernous malformations are the most common CNS manifestations (Fig. 12). Dural arteriovenous fistulas (AVFs) may be present. Commonly, CNS findings may relate to sequela of systemic AVMs, particularly embolic strokes and abscesses (Fig. 13).
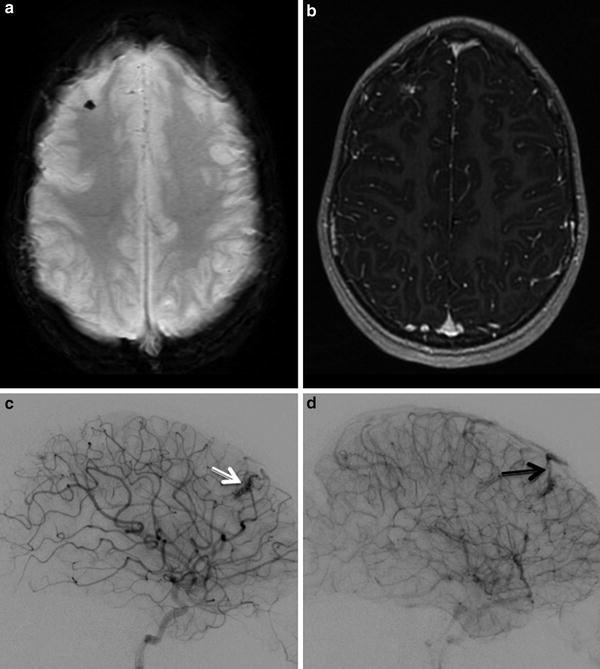
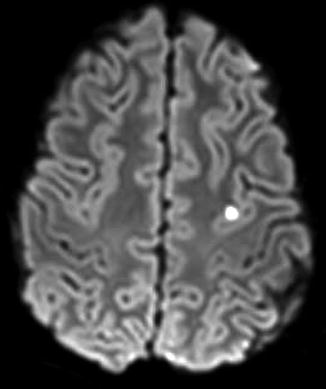
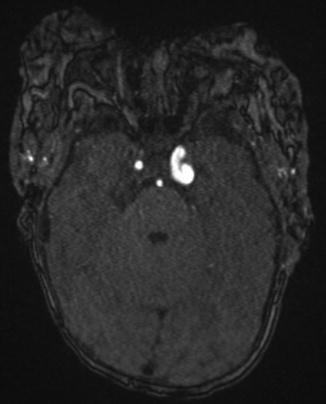
Fig. 12
Small AVM in a child with HHT. (a). Axial SWI shows blooming artifact in right frontal lobe. (b). Post contrast T1 weighted image shows associated focal enhancement. (c). Arterial phase injection of right ICA showing the right frontal AVM (white arrow). (d). Draining vein of AVM seen on subsequent image (black arrow)
Fig. 13
DWI shows a punctate embolic stroke in a teenager with HHT
Gene mutations have been identified, allowing subclassification of HHT. The most frequent mutation is HHT type 1 (HHT1) caused by mutation in ENG gene (chromosome 9q34). HHT2 has been mapped to ALK1 gene (12q), HHT3 (5q31), AND HHT4 (7p14), and juvenile polyposis/HHT syndrome has been linked to mutation in SMAD4 gene (18q21).
HHT patients often present in childhood, with clinically significant epistaxis (most common), evaluation of mucosal or skin telangiectasias, GI bleeding, or visceral angiodysplasias (Fig. 14). Screening for cerebral AVMs is often performed in patients with genetic or clinical diagnosis of HHT. Neurologic symptoms are occasionally the presentation of HHT, typically from embolic strokes or abscesses from pulmonary AVMs, which are a more typical manifestation. Cerebral AVMs are less common, but are more typical in ENG mutations than other known mutations.
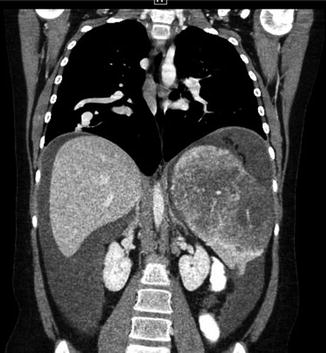
Fig. 14
Same patient as Fig. 13. Teenager with HHT. Coronal CT image demonstrates right-sided pulmonary AVM and angiodysplasia within the spleen. Hemorrhagic fluid is seen in the abdomen
Imaging for HHT patients typically includes workup and screening with MRI and MRA. Addition of susceptibility weighted imaging (SWI) or gradient echo sequences will maximize sensitivity to blood products. CTA is a useful tool, however, in the era of limiting pediatric exposure to ionizing radiation (As Low As Reasonably Achievable- ALARA principle), the use of MRA has increased. There are occasions when small AVMs are not well seen on MRA and may only be seen on digital subtraction angiography (Fig. 15), which is still considered the gold standard for diagnosis and also allows potential for treatment.
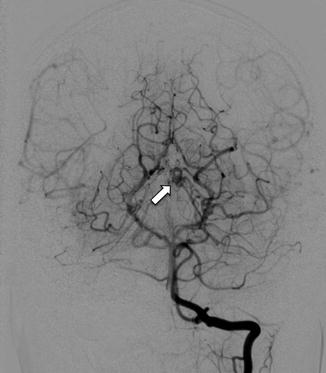
Fig. 15
Left vertebral injection in a teenage HHT patient showing a left thalamic AVM supplied by a PCA branch (arrow). This was not identified on conventional MRI/MRA
Wyburn-Mason
Wyburn-Mason syndrome (also known as Bonnet-Dechaume-Blanc syndrome or retinoencephalofacial angiomatosis) is classically described as the presence of an AVM of the visual pathway (typically retinal or orbital AVM), ipsilateral midbrain AVM and often an ipsilateral facial angioma. Wyburn-Mason studied patients with retinal AVMs and found a strong association with concomitant intracranial AVMs. The current classification does not necessitate all three findings of the classical description. The embryology is thought to relate to a fetal abnormality of the primitive mesoderm before 7 weeks gestational age causing persistence of early fetal vascular tissues shared by the developing optic cup and anterior neural tube. Abnormal vascular development leads to anomalous communications between arteries and veins, thus AVM formation. While midbrain AVMs are classically described, hypothalamic/thalamic and basal ganglia AVMs are also common. The ipsilateral facial angioma is only seen in 30–50 % of patients classified with Wyburn-Mason.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


