Disorders of Brain Development
Thierry A.G.M. Huisman
Andrea Poretti
Central Nervous System Embryogenesis
In order to understand the classification and pathogenesis of developmental disorders of the central nervous system (CNS) shown on imaging, it is useful to first review the fundamental steps in embryogenesis of the CNS.
On day 1, fertilization of an egg by a sperm results in the zygote. The zygote undergoes mitotic division, which results in daughter cells called blastomeres. Approximately 3 days after fertilization, at the 12- to 16-cell stage, the conceptus is termed the morula. Approximately 4 days after fertilization, at the 50- to 60-cell stage, the conceptus is termed the blastula and has divided into two components: an inner cell mass called the embryoblast, and an outer cell mass called the trophoblast that gives rise to part of the placenta.
During the second week of gestation, the embryoblast (inner cell mass) becomes bilaminar. The lamina of cells that faces the amniotic cavity is termed the epiblast, and the lamina of cells that faces the yolk sac is called the hypoblast. By the second week, the conceptus is composed of two layers.
During the third week of gestation, the conceptus becomes trilaminar, and the process by which the bilaminar embryo becomes trilaminar is termed gastrulation. Conveniently, one can remember that during the third week, the conceptus is composed of three layers. This process of conversion begins with the appearance of a thick, linear band along the dorsal caudal surface of the epiblast termed the primitive streak. Epiblast cells accumulate along the primitive streak, which results in cranial elongation of the streak. Epiblast cells begin to migrate between the epiblast and the hypoblast. At this time of migration, the embryo is considered trilaminar: the epiblast is renamed the ectoderm, the migrated mesenchymal epiblastic cells are termed the mesoderm, and the hypoblast is renamed the endoderm. While the primitive streak is developing, it thickens at its cephalic end to form a structure called the Henson’s node. A portion of the invaginating cells will remain in the midline and migrate along the craniocaudal axis of the primitive streak to form the notochordal process. After resorption of the floor of the notochordal process, the resulting prochordal plate will transform into the definitive notochord (at 20 days).
The notochord defines the primitive axis and skeleton of the embryo and will eventually be replaced by the vertebral column. It extends throughout the entire embryo and reaches as far as the level of the future midbrain, where it ends in the region of the future dorsum sella. Most importantly, the notochord induces the transformation of the overlying ectoderm into neuroectoderm with formation of the neural plate. The notochord secretes a protein called sonic hedgehog (SHH) which plays a critical role in signaling the development of motoneurons. Between days 18 and 20, the neural plate transforms into a neural groove which starts to close into a neural tube at day 21 of gestation (1).
While the neural tube is closing, the neuroectoderm progressively detaches from the adjacent surface ectoderm and will “dive” into the space between ectoderm and endoderm. The adjacent surface ectoderm will close dorsally to the neural tube. During the fourth week of gestation, the neural tube bares two open ends: the rostral/anterior neuropore and the cauda/posterior neuropore. The anterior and posterior neuropores will close at day 25 of gestation (Fig. 4.1), and provides a summary glimpse of the neural tube and cell migrations that occur around 24 to 25 days gestation. After closure of the rostral neuropore, the cranial end of the neural tube undergoes segmentation into neuromeres and rhombomeres. The neuromeres give rise to the prosencephalon (forebrain), which further divides into the telencephalon (cerebral hemispheres) and the diencephalon. The rhombomeres give rise to the mesencephalon (midbrain) and the rhombencephalon (hindbrain). Simultaneously, the spinal cord and paraspinal tissues will develop. Cells at the border of the neuroectoderm and ectoderm will detach to form the neural crests. The neural crests subsequently fragments and give rise to the primordial of the ganglia which again give rise to the sensory innervations. The corresponding level of the neural tube and later spinal cord furnishes the motor innervations. A somite plate develops on each side of the neural tube which also becomes segmented (somite). At the end of the fifth week of gestation, 42 pairs of somites are noted. The somites will develop a central cavity; the internal side will give rise to the sclerotome which again migrate toward the notochord and will become the vertebral primordial. Cells include fibro-, chondro-, and osteoblasts. The part of the somite which remains in place will become the dermomyotomes. The dermomyotomes are subsequently divided into dermatomes and myotomes. The myotomes give rise to the vertebral muscles. The notochord regresses at the level of the vertebral bodies but persists at the level of the intervertebral discs and will become the nucleus pulposus. The paraxial musculature derived from the somites remains segmental, allowing the musculature to bridge from one vertebral body to the next. The spinal nerves remain segmental and consequently leave the spinal canal between the intervertebral foramina.
In the further development of the brain, multiple, complex, interacting and programmed processes guide and determine the migration of the precursors of the neurons from, for example, the germinal matrix to the central and cortical gray matter. Subsequent intracortical migration and networking will determine the thickness, complexity, and shape of the internal and superficial architecture of the cortex. Many functional networks and
connections develop. Finally, progressing white matter myelination is observed in well-determined sequences. The development of the fetal brain does not end with birth but continues well into the first and partially second decade of postnatal life.
connections develop. Finally, progressing white matter myelination is observed in well-determined sequences. The development of the fetal brain does not end with birth but continues well into the first and partially second decade of postnatal life.
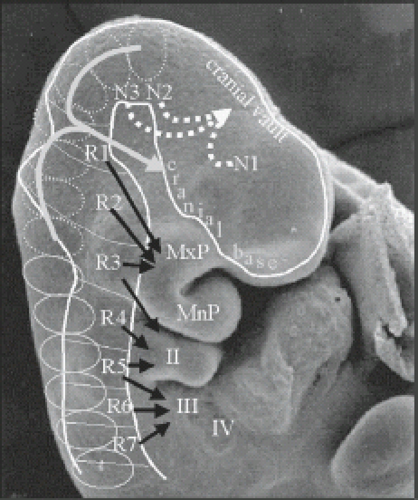 FIGURE 4.1 The neural tube at 24 to 25 days gestation. Scanning electron micrograph of a 10-day-old mouse embryo (equivalent human age 29 days). The neural tube (white outline), somites (ovals), pharyngeal arches, and neural crest cell migration are depicted schematically. Following closure of the anterior neuropore at 24 days gestation, the cranial neural tube undergoes segmentation: the three most cranial segments are termed neuromeres: N1 corresponds to the prosencephalon, and N2 and N3 to the mesencephalon. The next eight segments are termed rhombomeres, R1 to R8, and comprise the rhombencephalon. At 19 to 21 days gestation, the paraxial mesoderm condenses around the neural tube into cuboidal bodies termed somites. The seven most rostral somites are less well defined and are termed somitomeres (dashed ovals). By 29 days gestation, the four pharyngeal arches have appeared as surface elevations along the primitive oral cavity and pharynx. The maxillary prominence (MxP) and the mandibular prominence (MnP) comprise the first pharyngeal arch. The second (II), third (III), and fourth (IV) pharyngeal arches may be seen at this age. Neural crest cells form from surface ectoderm at the dorsal crests of the neural tube. There is an anatomic registration between the segments of the neural tube and neural crest cell migration, giving rise to the craniofacial primordia, depicted here as migrational streams (arrows). Neural crest cell migration from N1, N2, and N3 (dashed white arrows) gives rise to the cranial vault and calvaria (membranous neurocranium). Neural crest cell migration from the rhombomeres (black arrows) heralds the formation of the pharyngeal arches and gives rise to formation of the viscerocranium (face, jaws, middle ears). The cranial base (cartilaginous neurocranium) originates from the paraxial mesoderm (curved gray arrows). (Courtesy of K. Sulik, University of North Carolina, Chapel Hill, NC.) |
The reader is referred to neuroembryologic texts for a more comprehensive review of CNS embryogenesis (1).
Classification of CNS Malformations
Over the past two decades, significant progress in pre- and postnatal neuroimaging techniques, development of next-generation genetic sequencing, and animal model research has allowed advancement of the correct definition/classification of congenital brain abnormalities and, as a consequence, a better understanding of their pathogenesis. Indeed, classifications of congenital brain abnormalities have been proposed based upon neuroimaging, molecular genetics, or developmental biology (2,3,4).
Neuroimaging plays a key role in the primary diagnosis of congenital brain abnormalities. Accurate diagnoses of these complex abnormalities are of paramount significance for three primary reasons: (1) to determine inheritance pattern and risk of recurrence, (2) to document involvement of other systems (e.g., kidneys and liver in Joubert syndrome [JS]), and (3) to assist with understanding prognostic implications for the child and family. Additionally, the neuroimaging findings may allow the definition of subphenotypes within a group of congenital brain anomalies and establish correlations between the neuroimaging phenotype and genotype (e.g., in lissencephaly [LIS]).
The first question that has to be answered in any diagnostic workup of an anomalous pediatric brain is if the encountered findings are resulting from an inherited (genetic) versus an acquired (disruptive) cause.
A malformation is defined as a congenital morphologic anomaly of a single organ or body part due to an alteration of the primary developmental program caused by a genetic defect (5). Gene mutations causing malformations may be “de novo” or be inherited following different patterns that imply a different recurrence risk for further offspring. Typical examples include a syndromal corpus callosum agenesis, holoprosencephaly (HPE), JS, or rhombencephalosynapsis (RES), to mention a few.
A disruption is defined as a congenital morphologic anomaly due to the breakdown of a brain structure that had a normal developmental potential or was initially, intrauterine well developed (5). Disruptive causes include, for example, prenatal infection, hemorrhage, or ischemia. The etiology of prenatal hemorrhages is manifold and includes both maternal (e.g., trauma, sepsis, preeclampsia, and drugs abuse) and fetal (e.g., vascular malformations, congenital tumors, and alloimmune thrombocytopenia) causes. Typical examples of prenatal disruptions affecting the supratentorial brain include hydranencephaly or hemi-hydranencephaly (Fig. 4.2A), twin disruption sequence (Fig. 4.2B), and the fetal brain disruption (FBD)-like phenotype. Disruptions are acquired lesions with very low recurrence risk. However,
a genetic predisposition to disruptive lesions may be present resulting in an overlap between both etiologies. Dominant mutations in COL4A1 lead to change of the basal membrane of capillaries resulting in microangiopathy (Fig. 4.3). Within the brain, the microangiopathy may lead to hemorrhages and/or ischemias and result in porencephaly or unilateral cerebellar hypoplasia (6). In addition, recently homozygous mutations in NED1 have been shown to cause severe microcephaly, agenesis of the corpus callosum, scalp rugae, and the FBD-like phenotype.
a genetic predisposition to disruptive lesions may be present resulting in an overlap between both etiologies. Dominant mutations in COL4A1 lead to change of the basal membrane of capillaries resulting in microangiopathy (Fig. 4.3). Within the brain, the microangiopathy may lead to hemorrhages and/or ischemias and result in porencephaly or unilateral cerebellar hypoplasia (6). In addition, recently homozygous mutations in NED1 have been shown to cause severe microcephaly, agenesis of the corpus callosum, scalp rugae, and the FBD-like phenotype.
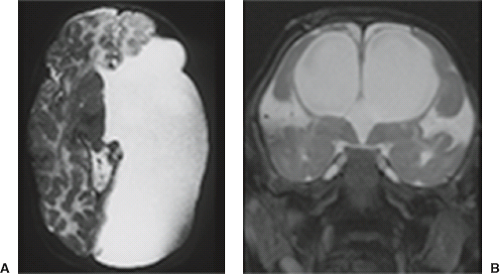 FIGURE 4.2 Examples of prenatal disruptions affecting the supratentorial brain. A: Axial T2-weighted image of a child with left hemihydranencephaly shows almost complete absence of the left cerebral hemisphere with the exception of part of the left frontal lobe matching the vascular territory of the left anterior cerebral artery. B: Coronal T2-weighted image of a neonate after fetal twin–twin transfusion reveals severe symmetric brain tissue loss and marked, secondary ventriculomegaly. |
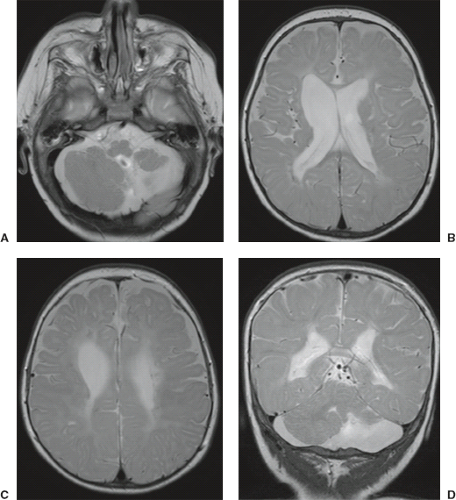 FIGURE 4.3 An 8-month-old child with bilateral spastic cerebral palsy and COL4A1 mutation. (A–C) Axial and (D) coronal T2-weighted images show unilateral left cerebellar hypoplasia as the results of a prenatal disruptive lesion, bilateral T2-hyperintense signal of the periventricular white matter, and mild ventriculomegaly. |
There are many old and new classification systems. Some are based upon historic discoveries and reports, while others are based upon the predominant clinical presentation, neuropathologic findings, genetic and/or molecular data, or on neuroimaging studies. Any classification should be comprehensive and practical, but most importantly correct in order to provide relevant information about prognosis, treatment options, and likelihood of recurrence.
Neuroimaging has long served as a noninvasive alternative to neuropathology to determine or confirm a clinically suspected diagnosis. Today, neuroimaging has evolved from a confirmatory diagnostic tool into a modality in which the correct identification and classification of morphologic and microstructural neuroimaging findings initiate the targeted search for a genetic basis of a brain anomaly (e.g., the detection of an anomalous decussation of the superior cerebellar peduncles (SCP) in children with a so-called tectocerebellar dysraphia suggested that this malformation is related to the well-known JS). In this chapter, we have opted for an updated classification based on the neuroimaging pattern, one that is usable in daily clinical work. This classification also incorporates our current knowledge about macro- and microscopic embryology, axonal guidance and signaling, genetics, and microstructural/functional neuroimaging findings.
Anomalies of Dorsal Prosencephalon Development
Anomalies of the Corpus Callosum and Other Cerebral Commissures
Definition
The corpus callosum may be completely absent (agenesis) or partially formed (hypogenesis). Additionally, malformations of the other commissures (anterior and hippocampal commissures) may be associated with corpus callosum agenesis or hypogenesis leading to a complex spectrum of various commissural disorders (7). Moreover, in addition to anomalies of the other telencephalic commissures, anomalies of the corpus callosum are often associated with additional cerebral or cerebellar abnormalities such as interhemispheric
cysts, malformations of the cortical development, cerebellar dysgenesis, cephaloceles, or hypothalamic anomalies.
cysts, malformations of the cortical development, cerebellar dysgenesis, cephaloceles, or hypothalamic anomalies.
Clinical Features
Although asymptomatic isolated callosal agenesis has been reported, the vast majority of affected patients present with seizures, developmental delay, cognitive impairment, or hypothalamic dysfunction. Callosal anomalies may be isolated or part of many complex syndromes such as Aicardi syndrome, fetal alcohol syndrome, Chiari II malformation, Dandy–Walker malformation (DWM), nonketotic hyperglycinemia, or pyruvate dehydrogenase deficiency (8). The most frequent is probably the Aicardi syndrome, a likely X-linked dominant disorder characterized by the triad of callosal agenesis, infantile spasms, and chorioretinal lacunae. Typical additional structural brain abnormalities include polymicrogyria (PMG) predominantly frontal and perisylvian, periventricular or subcortical nodular heterotopias, intracranial cysts, enlargement of the tectum in about the half of the patients, and cerebellar anomalies (9). As can be expected, the associated findings are often the main cause of clinical symptoms.
Pathogenesis
Many scientists have studied the development of the corpus callosum and many misconceptions resulted. The false concept that the corpus callosum develops from anterior (genu) to posterior (splenium) and that the rostrum appears at the very end of the development survived for many decades. Scientists including Jim Barkovich and Charles Raybaud as well as many others have authored several concept manuscripts detailing the complex development of the corpus callosum. We would like to defer to the respective articles (7). Most important is that each neuroradiologist is familiar with the wide variation in shape, length, and thickness of the normal corpus callosum, and that he/she knows to accurately differentiate between a corpus callosum malformation (isolated versus syndromal), corpus callosum destruction (e.g., adjacent hemispheric infarction), and a high-grade corpus callosum thinning (high-grade hydrocephalus). The functional prognosis and outcome vary accordingly.
Imaging Findings
On conventional T1- and T2-weighted MRI, the main finding in callosal anomalies is absent or hypoplastic corpus callosum itself (Figs. 4.4 and 4.5). Most frequently, the posterior part and inferior genu and rostrum are absent. However, many degrees and variations of hypogenesis/agenesis may be present. Parallel coursing, mildly lateralized and separated lateral ventricles, colpocephaly, and upward extension of the third ventricle into the interhemispheric fissure between the lateral ventricles are characteristic neuroimaging findings of callosal agenesis (Figs. 4.6 and 4.7) (7).
Other MR findings in the absence of the corpus callosum include lack of definition or inversion of the cingulate gyrus (Fig. 4.8), crescent-shaped lateral ventricles (caused by an impression upon the medial walls of the ventricles by the medially positioned bundles of Probst), separated leaves of the septum pellucidum, malrotation of the hippocampi, and absence of the inferior cingulum (Figs. 4.4 and 4.6) (7). The Probst bundles refer to the white matter tracts that would normally have crossed the interhemispheric fissure within the corpus callosum but instead run parallel to the interhemispheric fissure in an anterior to posterior extension (Fig. 4.9). Due to the lack of the inversion of the cingulate gyrus, the cingulate sulcus is absent/shallow and, consequently, the sulci along the mesial hemispheric surface appear to radiate toward the third ventricle.
Other MR findings in the absence of the corpus callosum include lack of definition or inversion of the cingulate gyrus (Fig. 4.8), crescent-shaped lateral ventricles (caused by an impression upon the medial walls of the ventricles by the medially positioned bundles of Probst), separated leaves of the septum pellucidum, malrotation of the hippocampi, and absence of the inferior cingulum (Figs. 4.4 and 4.6) (7). The Probst bundles refer to the white matter tracts that would normally have crossed the interhemispheric fissure within the corpus callosum but instead run parallel to the interhemispheric fissure in an anterior to posterior extension (Fig. 4.9). Due to the lack of the inversion of the cingulate gyrus, the cingulate sulcus is absent/shallow and, consequently, the sulci along the mesial hemispheric surface appear to radiate toward the third ventricle.
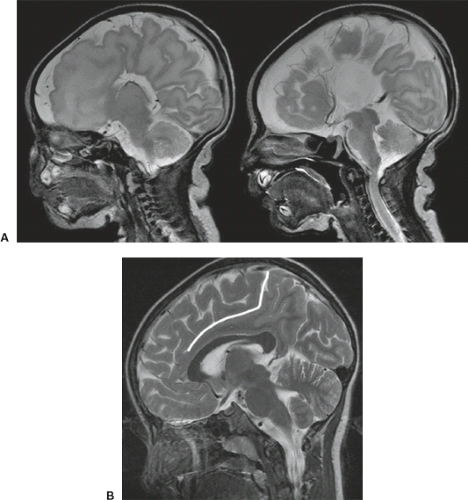 FIGURE 4.4 Complete agenesis of the corpus callosum. A,B: Midsagittal T2-weighted images show complete absence of the corpus callosum, cingulate gyrus, and cingulate sulcus, radiation of the medial hemispheric sulci toward the cerebral hilum, and expansion of the roof of the third ventricle, which is not maintained by the commissural plate. C: Normal midsagittal T2-weighted image of a healthy child for comparison (the white line delineates the cingulate sulcus). |
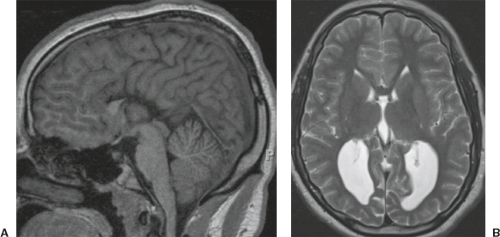 FIGURE 4.5 Partial agenesis of the corpus callosum. A: Midsagittal T1-weighted image shows a rudiment of the corpus callosum close to the anterior commissure, small posterior fornix, and hippocampal commissures, which are attached to the splenium. This means that the commissural plate is virtually complete even though it is very short. B: Axial T2-weighted image reveals colpocephaly. |
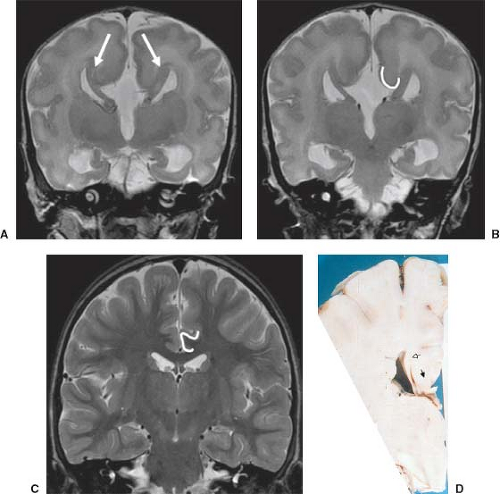 FIGURE 4.6 Complete agenesis of the corpus callosum. A,B: Coronal T2-weighted images show widely separation of the lateral ventricles, that have a “Texas longhorn” configuration and contain the heavily myelinated bundles of Probst (A, white arrows); the cingulate gyri are not inverted (the white line delineates the left cingulate gyrus); the third ventricle is extended upwards into the interhemispheric fissure between the lateral ventricles; there is absence of hippocampal rotation. C: Normal coronal T2-weighted image of a healthy child for comparison (the white line delineates the left cingulate gyrus, which is physiologically inverted). D: Coronal necropsy specimen shows a concave frontal horn due to a combination of the Probst callosal bundle (open arrows) and an everted cingulate gyrus (closed arrows). |
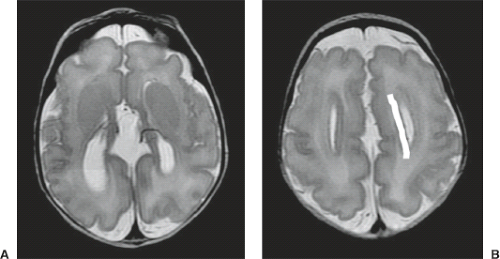 FIGURE 4.7 Complete agenesis of the corpus callosum. A,B: Axial T2-weighted images reveal extension of the third ventricle between the cerebral hemispheres, parallel course of the lateral ventricles (white line) with medially located heavily myelinated bundles of Probst, and colpocephaly. |
Occasionally, an interhemispheric cyst is seen in close proximity to the high-riding third ventricle (Fig. 4.10). Although arachnoid cysts can occur in the interhemispheric fissure in association with agenesis of the corpus callosum, more commonly, this cerebrospinal fluid (CSF) collection is lined by
ependymal cells. It may communicate with the third ventricle or one or both of the lateral ventricles.
ependymal cells. It may communicate with the third ventricle or one or both of the lateral ventricles.
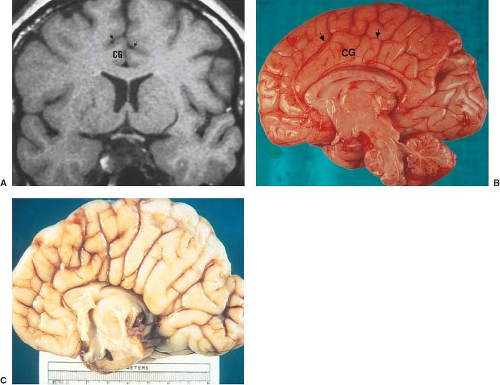 FIGURE 4.8 Normal cingulate gyrus and cingulate sulcus formation. A: Spin-echo 600/20 image of normally inverted cingulate gyri. B: Sagittal necropsy specimen, showing normal cingulate gyrus and sulcus. C: Sagittal necropsy specimen in posterior agenesis of the corpus callosum. When the corpus callosum forms, one consequence of the normal crossing of the callosal fibers is inversion of the cingulate gyri (CG in A,B) with resultant formation of the cingulate sulci (A,B, arrows). In callosal agenesis (C), the lack of formation of a well-defined cingulate sulcus results in mesial hemispheric sulci radiating in an uninterrupted fashion to the roof of the third ventricle in the region of the absent corpus callosum. |
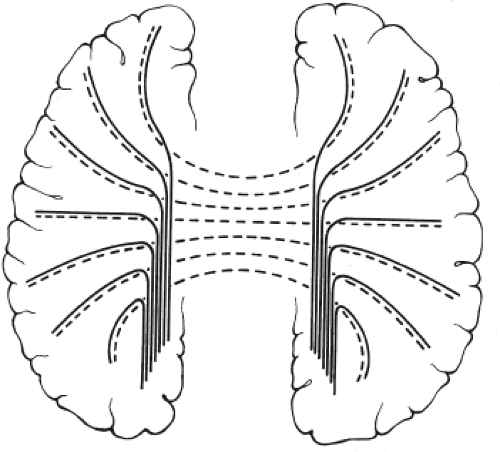 FIGURE 4.9 The formation of the lateral callosal bundles (of Probst). As a result of a lack of normal formation of the commissural plate, axons from the cerebral hemispheres do not cross the midline. Instead, on reaching the medial hemispheric wall, the fibers turn to course parallel to the interhemispheric fissure. The fibers run parallel to and indent the medial walls of the lateral ventricles. Broken lines represent normal callosal fibers. Solid lines represent fibers that fail to cross the midline and instead form the lateral callosal bundles. |
Because the corpus callosum is only one of the three major commissures next to the anterior and hippocampal commissures, these additional commissures should be carefully evaluated for associated anomalies.
Furthermore, in syndromal corpus callosum malformations like, for example, Aicardi syndrome, additional brain anomalies like migrational abnormalities, disorders of cortical organization, and myelination abnormalities should be excluded (Fig. 4.11).
Diffusion tensor imaging (DTI) and fiber tractography (FT) allow to better understand and explore corpus callosum malformations. In patients with agenesis of the corpus callosum, next to the lack of the left–right crossing commissural fibers, the most obvious DTI finding is the presence of the bundles of Probst as large, anterior–posterior oriented (green on color-coded Fractional anisotropy (FA) maps), intrahemispheric, heterotopic white matter tracts coursing along the medial and superior wall of the lateral ventricles (Fig. 4.12). The bundles of Probst may already be depicted by fetal DTI (Fig. 4.13). The bundles of Probst have been shown to connect anterior regions of the hemisphere with more posterior parts exhibiting at least a partially topographic organization. The bundles of Probst are contained in the separated leaves of the septum pellucidum and appear to be continuous with the superior cingulum and fornices. In patients with agenesis of the corpus callosum, a DTI study showed an abnormal microstructure of the right ventral cingulum bundle (as reduced FA values) and bilateral reduction in length and volume. Abnormalities of the cingulum may mirror the observed clinical
deficits in executive function and social–emotional processing in patients with callosal agenesis. In patients with agenesis of the corpus callosum, DTI also showed that the fornices are dysplastic and widely separated. In partial agenesis of the corpus callosum, DTI and FT typically show crossing fibers in the partially developed parts of the corpus callosum. Two DTI studies focused on callosal connectivity in partial agenesis of the corpus callosum. The first study showed a relatively consistent pattern of anteriorly located callosal fragments with primarily homotopic frontal connectivity. The study also revealed heterotopic callosal fibers that coursed from right anterior frontal lobe to left occipitotemporal lobes. The second study demonstrated a greater diversity of partial callosal connectivity, including a number of heterotopic tracts that are absent in normally developed, healthy subjects. The patterns of the callosal connections could not be predicted from the appearance of the callosal fragments on conventional MRI. These studies suggest that partial agenesis of the corpus callosum may result from more complex processes than just arrested callosal development.
deficits in executive function and social–emotional processing in patients with callosal agenesis. In patients with agenesis of the corpus callosum, DTI also showed that the fornices are dysplastic and widely separated. In partial agenesis of the corpus callosum, DTI and FT typically show crossing fibers in the partially developed parts of the corpus callosum. Two DTI studies focused on callosal connectivity in partial agenesis of the corpus callosum. The first study showed a relatively consistent pattern of anteriorly located callosal fragments with primarily homotopic frontal connectivity. The study also revealed heterotopic callosal fibers that coursed from right anterior frontal lobe to left occipitotemporal lobes. The second study demonstrated a greater diversity of partial callosal connectivity, including a number of heterotopic tracts that are absent in normally developed, healthy subjects. The patterns of the callosal connections could not be predicted from the appearance of the callosal fragments on conventional MRI. These studies suggest that partial agenesis of the corpus callosum may result from more complex processes than just arrested callosal development.
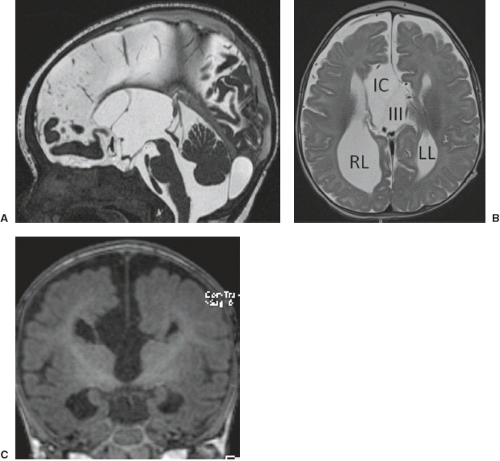 FIGURE 4.10 Complete agenesis of the corpus callosum with interhemispheric cysts. A: Sagittal CISS image shows complete absence of the corpus callosum. B: Axial T2-weighted image reveals a right interhemispheric cyst (IC, interhemispheric cyst; III, third ventricle; LL, left lateral ventricle; RL, right lateral ventricle). C: Coronal T1-weighted image shows a “Texas longhorn” configuration of the lateral ventricles, an upwards extension of the third ventricle, a right interhemispheric cyst, and lack of rotation of the hippocampi. |
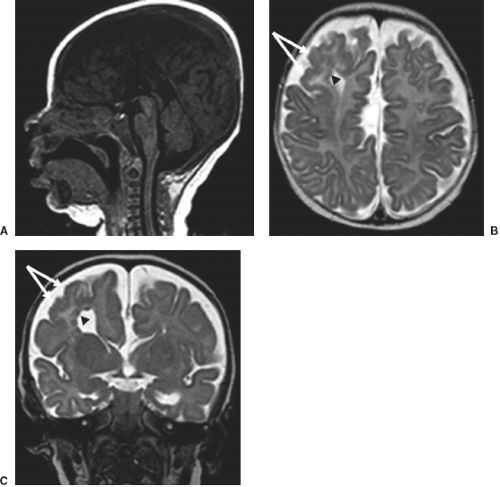 FIGURE 4.11 Aicardi syndrome. A: Midsagittal T1-weighted image shows absence of the corpus callosum and septum pellucidum. B: Axial T2-weighted image reveals asymmetric dilatation and deformity of the anterior horn of the right lateral ventricle with subependymal, nodular heterotopia (short arrows) and polymicrogyria of the overlying cortex (long arrows). C: Coronal T2-weighted image shows absence of the corpus callosum and asymmetric dilatation and deformity of the anterior horn of the right lateral ventricle with subependymal, nodular heterotopia (short arrows) and polymicrogyria of the overlying cortex (long arrows). (Reprinted with permission from Hergan B, Atar OD, Poretti A, et al. Serial fetal MRI for the diagnosis of Aicardi syndrome. Neuroradiol J 2013;26:380–384.) |
 FIGURE 4.12 Complete corpus callosum agenesis. (A) Axial and (B) coronal color-coded FA maps show the bundles of Probst as large, longitudinally oriented (green on color-coded FA maps) white matter tracts that are running along the medial and superior wall of the lateral ventricles. C: Tractography superimposed on an axial T2-weighted image confirms the bundles of Probst. (Reprinted with permission from Poretti A, Meoded A, Rossi A, et al. Diffusion tensor imaging and fiber tractography in brain malformations. Pediatr Radiol 2013;43:28–54.) |
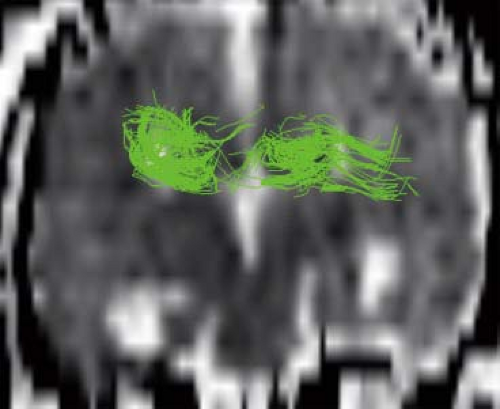 FIGURE 4.13 Complete corpus callosum agenesis. Fetal tractography superimposed on a coronal T2-weighted image shows the Probst bundles as thick, parallel bundles coursing medially to the lateral ventricles. (Reprinted with permission from Meoded A, Poretti A, Tekes A, et al. Prenatal MR diffusion tractography in a fetus with complete corpus callosum agenesis. Neuropediatrics 2011;42:122–123.) |
Finally, it should be mentioned that a corpus callosum malformation can easily be detected by neonatal head ultrasound examinations (Fig. 4.14). However, the associated anomalies including migrational abnormalities, malrotation of the hippocampi or, for example, a concomitant absence of the anterior, or hippocampal commissure cannot be excluded by ultrasound studies.
Malformation of Cerebral Cortical Development
The development of the human cerebral cortex is a highly complex process that is regulated by a high number of genes and
can be divided into three broad and overlapping steps: (1) neural stem cell proliferation and cell-type differentiation, (2) neuronal migration, and (3) cortical organization and connectivity (4). Any abnormality that interferes with one or more of these processes may result in a malformation of the cortical development. These abnormalities may include (1) gene mutations causing a primary malformation, (2) destructive events (e.g., infection or hemorrhage) causing a disruption, and (3) exogenous toxins (e.g., drugs or alcohol from maternal ingestion, or endogenous toxins from metabolic disorders such as Zellweger syndrome).
can be divided into three broad and overlapping steps: (1) neural stem cell proliferation and cell-type differentiation, (2) neuronal migration, and (3) cortical organization and connectivity (4). Any abnormality that interferes with one or more of these processes may result in a malformation of the cortical development. These abnormalities may include (1) gene mutations causing a primary malformation, (2) destructive events (e.g., infection or hemorrhage) causing a disruption, and (3) exogenous toxins (e.g., drugs or alcohol from maternal ingestion, or endogenous toxins from metabolic disorders such as Zellweger syndrome).
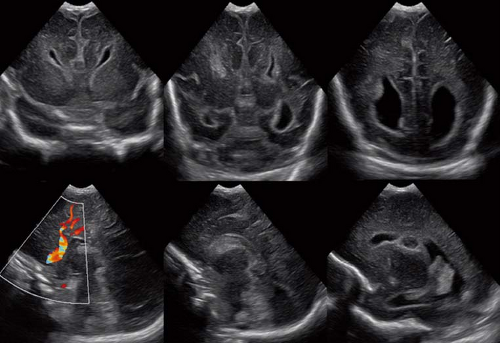 FIGURE 4.14 Complete corpus callosum agenesis. Coronal and sagittal head ultrasound images show the characteristic “Texas longhorn” configuration of the lateral ventricles, radiation of the mesial sulci from the region of the third ventricle, and colpocephaly. Incidental note is made of a resolving right germinal matrix hemorrhage grade 2. (Reprinted with permission from Orman G et al., J Neuroimaging, 2015;25:31–55.) |
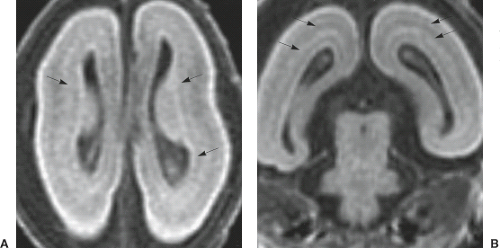 FIGURE 4.15 Fetal brain, 14 weeks gestation. (A) Axial and (B) coronal images from three-dimensional gradient-echo sequence obtained on a fetal anatomy specimen show smooth brain without sulcal development. Note germinal matrix (A, arrows) and normal waves of neuronal migration (B, arrows). |
After a short introduction about normal embryology of the cerebral cortex, we will discuss the various malformations of cerebral cortical formation. We will classify them into three groups: (1) malformations secondary to abnormal stem cell proliferation or apoptosis, (2) malformations secondary to abnormal neuronal migration, and (3) malformations secondary to abnormal late migration and cortical organization.
Normal Development of the Cerebral Cortex
A review of normal cortical formation is useful for understanding the malformations of cortical development. The developing prosencephalon consists of two parts: (1) a thin dorsal portion (also called pallium or dorsal germinal zone) that will form the cortex and hippocampus, and (2) a thick basal portion (also called subpallium or ventral germinal zone) that will form the basal ganglia. More in detail, the basal ganglia derive from two primordia that form as bulges or eminences along the lateral ventricle: the primordium of the globus pallidus, termed the medial ganglionic eminence, and the primordium of the corpus striatum, termed the lateral ganglionic eminence. A third eminence, termed the caudal ganglionic eminence, is believed to give rise to the amygdala. The cells that proliferate within the ventral germinal zone include interneurons that express the neurotransmitter γ-aminobutyric acid (GABA) and have a tangential (nonradial) migration. The cells that proliferate within the dorsal germinal zone include projection neurons that express the neurotransmitter glutamate and have a radial migration to the cerebral cortex.
The traditional view of cerebral cortical formation has been termed the protomap hypothesis. According to this hypothesis, nearly all neuronal precursors migrate from the ventricular zone to the cortical plate guided by radial glial fibers (Figs. 4.15 and 4.16). From the ventricular zone of cell proliferation, known as the germinal matrix, cells begin to migrate centrifugally to form the cerebral cortex. Initial migrations begin during the eighth gestational week. A 1:1 or point-to-point correlation exists between the part of the germinal matrix in which certain neurons originate and the part or lamina of the cerebral cortex where the neurons come to rest (Fig. 4.17). This correlation is maintained largely by the presence of radially oriented glial fibers that span the hemisphere and act as scaffolding along which the neurons migrate (Fig. 4.18). After cell migration, according to the protomap hypothesis, the radial glial cells regress and form the mature glia.
The understanding of cortical formation, however, has considerably evolved since the protomap hypothesis. The radial
glia, for example, is now known to play a much greater role in neurogenesis and has been shown to be cortical neuronal as well as glial precursors.
glia, for example, is now known to play a much greater role in neurogenesis and has been shown to be cortical neuronal as well as glial precursors.
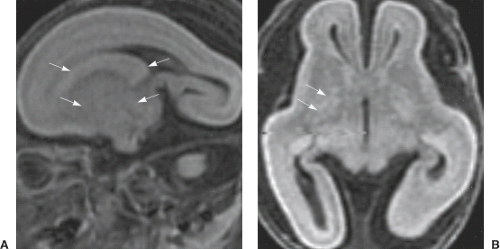 FIGURE 4.16 Fetal brain, 14 weeks gestation. (A) Sagittal and(B) axial images from three-dimensional gradient-echo sequence obtained on a fetal anatomy specimen. Early neuronal condensations of future caudate and lenticular nuclei can be seen (arrows). |
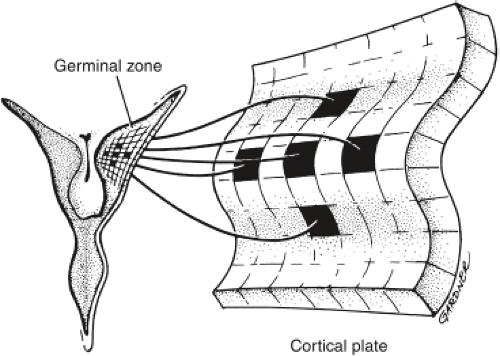 FIGURE 4.17 Schematic drawing illustrating the relationship of the germinal matrix to the developing cortical plate. A 1:1 correspondence exists between the site of cell proliferation in the germinal zone and its eventual resting place in the cortical plate. |
The choreography of neuronal migration, moreover, has been shown to be much more complex than the traditional centrifugal pattern. Precursor neurons of the piriform cortex have been shown to originate at the cortical–striatal boundary. These precursor neurons pass from the cortical–striatal boundary in an indirect, non–point-to-point fashion along a radial glial pathway known as the lateral cortical stream. Interneurons of the olfactory bulb, for example, arise in the subventricular zone and move along a rostral path termed the rostral migratory stream that does not involve radial glia. A large-scale ventral-to-dorsal tangential migration, moreover, has been shown to occur from the ganglionic eminences to the neocortex and accounts for much of the GABAergic interneurons that constitute 15% to 20% of all neocortical neurons. These migrational routes are summarized in Figure 4.19.
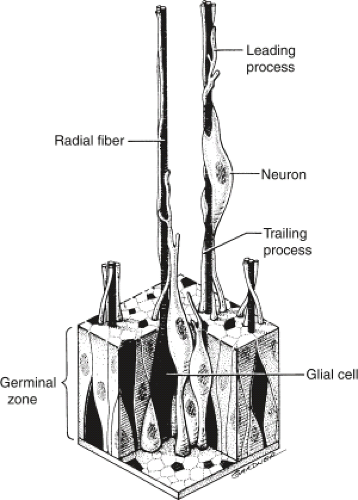 FIGURE 4.18 Drawing demonstrating the relationship of the migrating neurons to the radial glial cells. A migrating neuron is illustrated ascending the radial glial fiber. Damage to the radial glial fiber results in an arrest of cell migration. |
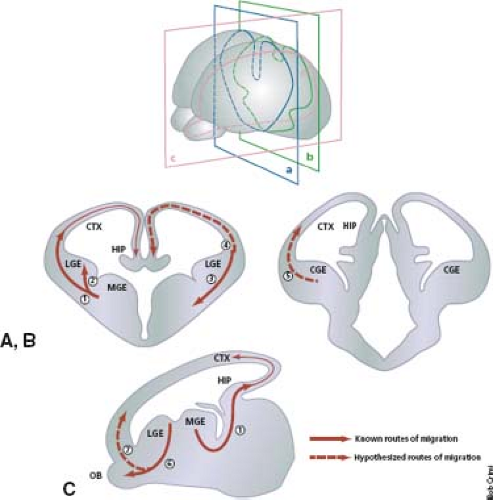 FIGURE 4.19 Known and putative routes of tangential cell migration. (A,B) Coronal and (C) sagittal sections of the telencephalon. The known routes of migration are depicted as solid arrows; putative routes are depicted by dashed arrows. (1) Medial ganglionic eminence (MGE) to neocortex (CTX) and hippocampus, (2) medial ganglionic eminence to lateral ganglionic eminence (LGE), (3) cortical–striatal boundary to ventrolateral telencephalon (lateral cortical stream), (4) LGE to neocortex and hippocampus, (5) caudal ganglionic eminence (CGE) to dorsal telencephalon, (6) LGE to olfactory bulb (rostral migratory stream), (7) retrobulbar region to marginal zone 75, 76. HIP, hippocampus. (From Corbin J, Nery S, Fishell G. Telencephalic cells take a tangent: non-radial migration in the mammalian forebrain. Nat Neurosci 2001;4(Suppl.):1177–1182, with permission.) |
Malformations Due to Abnormal Neuronal and Glial Proliferation or Apoptosis
Microcephaly
Definition
Microcephaly is defined by a head circumference that is less than two standard deviations under the norm for age, gender, and ethnicity (10,11). Microcephaly affects about 1 in 250,000 people and may be associated with normal or short stature, normal cortex or malformations of cortical development, and high or severely impaired neurologic and cognitive function. Microcephaly is usually divided into two main groups: (1) primary microcephaly with a genetic (malformative) etiology resulting in abnormal proliferation of neuronal precursors or apoptosis, and (2) secondary microcephaly, which results from late prenatal, perinatal, or postnatal injury.
In the 2012 updated classification of malformations of cortical development, Barkovich et al. (4) classify congenital microcephaly into eight different groups:
Microcephaly with severe intrauterine growth retardation and short stature. This group includes Seckel syndrome with mutations in ATR and microcephalic osteodysplastic primordial dwarfism syndrome (associated with mutations in multiple genes).
Microcephaly with variable short stature, moderate to severe developmental delay/intellectual disability, normal to thin cortex, simplified gyral pattern, and with/without callosal
hypogenesis. This groups includes Seckel syndrome due to mutations in CENPJ and CEP152.
Microcephaly with mildly short stature or normal growth, mild to moderate developmental delay/intellectual disability, normal or thin cortex, and with/without simplified gyral pattern, callosal hypogenesis, and/or focal periventricular nodular heterotopia (PNH). This group includes autosomal recessive primary microcephaly due to mutations in ASPM, MCPH1, CDKRAP5, and STIL.
Microcephaly with mildly short stature or normal growth, severe developmental delay/intellectual disability, variable cortical development with simplified gyral pattern or cortical dysgenesis, and with/without agenesis of the corpus callosum. This group includes autosomal recessive primary microcephaly due to mutations in PNKP, WDR62 (diffuse or asymmetric PMG), NDE1, and TBR2 (PMG and callosal agenesis).
Microcephaly with variable anomalies and with/without simplified gyral pattern, PNH, and/or cerebellar hypoplasia. This group includes patients with ARFGEF2 mutations (PNH).
Microcephaly with severe developmental delay/intellectual disability and evidence of degeneration with/without mild short stature, enlarged extra-axial spaces, callosal agenesis, and/or atypical cortical dysgenesis. This group includes Amish lethal microcephaly due to SLC25A19 mutations.
Microcephaly with LIS. This group includes Barth and Norman–Roberts syndromes.
Microcephaly with tissue loss and enlarged ventricles with/without cortical dysplasia and/or callosal agenesis. This group includes FBD and FDB-like sequences and familial “microhydranencephaly” due to MHAC mutations.
Clinical Features
The clinical presentation of microcephaly is heterogeneous (12). Cognitive impairment varies from moderate to severe. In children with moderate microcephaly, behavior is the predominant feature, while in children with severe microcephaly, seizures and spasticity are the main findings. A variety of craniofacial dysmorphic features may be present.
In terms of diagnosis, therapy, and prognosis, it is important to differentiate primary inherited microcephaly from progressive microcephaly due to a neurodegenerative disorder.
Pathogenesis and Genetics
The pathomechanisms of microcephaly are complex and multifactorial and include genetic and acquired causes. An increasing number of genes (mentioned above) have been associated with primary microcephaly. Mutated genes may cause abnormal mitotic microtubule spindle structure, numerical and structural abnormalities of the centrosome, altered cilia function, impaired DNA repair, DNA damage response signaling and DNA replication, along with attenuated cell cycle checkpoint proficiency (10). Many of these processes are highly interconnected.
Neuropathology and Imaging Findings
The cardinal feature in microcephaly is a small head and reduced craniofacial ratio. In addition, a simplified gyral pattern may be seen (Figs. 4.20 and 4.21). This pattern consists of a reduced number of gyri separated by abnormally shallow sulci.
Common associated abnormalities include foreshortened frontal lobes, mildly enlarged lateral ventricles, and a thin or even partially absent corpus callosum. Posterior fossa involvement in primary microcephaly was recently shown. PMG, PNH, and enlarged extra-axial spaces are less common.
Common associated abnormalities include foreshortened frontal lobes, mildly enlarged lateral ventricles, and a thin or even partially absent corpus callosum. Posterior fossa involvement in primary microcephaly was recently shown. PMG, PNH, and enlarged extra-axial spaces are less common.
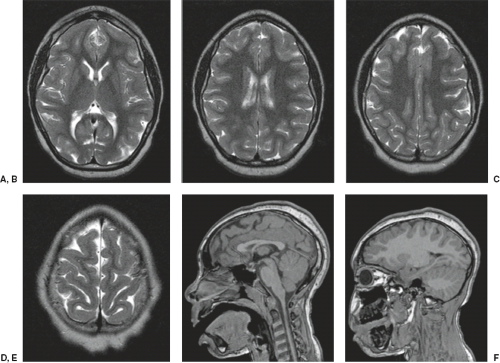 FIGURE 4.20 MR images of a 6-year-old boy with developmental delay and primary microcephaly. (A–D) Axial T2-weighted and (E,F) sagittal T1-weighted images show a mildly simplified gyral pattern including too few gyri and shallow sulci, normal myelination, and a normal thickness of cerebral cortex. |
 FIGURE 4.21 MR images of a 5-day-old male neonate with dysmorphic features, hypoplastic right heart, optic nerve hypoplasia, and severe primary microcephaly. (A,B) Sagittal and (C,D) axial T2-weighted images show an extremely simplified gyral pattern, a very thin corpus callosum, and disproportionally large cerebellum and brainstem compared to the cerebrum. |
Focal Cortical Dysplasia
Definition
Focal cortical dysplasia (FCD) is defined by the presence of abnormal neurons and glia within localized regions of the cerebral cortex. FCD is among the most common causes of epilepsy attributable to focal cerebral dysgenesis.
FCD is currently subdivided into four different groups (4):
Minor malformations of cortical development, which are characterized by normal cortical architecture and abundant ectopic neurons;
FCD type I, which are associated with architectural disturbances of the radial (type Ia) or tangential (type Ib) arrangement of cortical neurons;
FCD type II, which is characterized by more pronounced architectural abnormalities such as dysmorphic neurons (type IIa) and balloon cells (BCs) (type IIb);
FCD type III, which is associated with hippocampal sclerosis (type IIIa), glial or glioneuronal tumors (type IIIb), and vascular malformations (type IIIc).
FCD type II is the most prevalent type.
Clinical Features
Patients with FCD present with seizures that may be simple partial, complex partial, or secondarily generalized. Patients with FCD type II usually have extratemporal seizures that present at a younger age and have a higher frequency compared to patients with FCD type I. Developmental delay, intellectual disability, and focal neurologic deficits are unusual and only seen in patients with extensive FCD. The majority of the cases are sporadic; no familial recurrence.
Pathogenesis and Genetics
The pathogenesis of FCD is still largely unknown. An association was shown between FCD type IIb and expression of human papillomavirus 16 E6 oncoprotein in large BCs, but needs confirmation. The focal and variable nature of FCD type IIb and the pathologic similarities with tubers in tuberous sclerosis suggests that somatic mosaic mutations of genes involved in the mTOR pathway may be implicated in FCD.
Neuropathology and Imaging Findings
FCD typically exhibits abnormal cortical lamination, an indistinct cortical–white matter junction, and hypomyelination with astrogliosis of the adjacent white matter. Histologically, this form of FCD resembles the cortical tubers of tuberous sclerosis. In these lesions, the presence of abnormal BCs suggests abnormal cell proliferation or differentiation. In FCD, the histologic changes may be localized to the cortex and the immediate subcortical white matter but may, on occasion, extend from the pia to the ventricular surface. In some cases of FCD, the cortex is disorganized but lacks the BCs of the classic form, suggesting a postproliferation, postmigration abnormality of cortical organization.
FCD is rarely seen on CT and may not be visible even with high-quality MRI (e.g., mild malformations of cortical development and FCD type I). Subtle abnormalities in gyration (70%) (Fig. 4.22), cortical thickness (70%) (Fig. 4.22), and blurring of the gray–white matter junction (80%) (Fig. 4.23) are the most common feature and are best seen using thin-slice T1-weighted
images (13). The subcortical white matter may exhibit hyperintense signal on T2-weighted and fluid-attenuated inversion recovery (FLAIR), as well as hypointense signal on T1-weighted images as compared to white matter with mature myelination (Figs. 4.22 and 4.23). The white matter signal abnormality may be due to the presence of BCs or neurons within the dysplastic white matter or to abnormal or advanced myelination secondary to frequent seizures activity (14). In FCD without BCs, the lesions may be evident only as areas of blurring of the cortical gray–white matter junction. Cortical calcifications are occasionally demonstrated by CT. On imaging, FCD may be overt or extremely subtle and may occur in any lobe.
images (13). The subcortical white matter may exhibit hyperintense signal on T2-weighted and fluid-attenuated inversion recovery (FLAIR), as well as hypointense signal on T1-weighted images as compared to white matter with mature myelination (Figs. 4.22 and 4.23). The white matter signal abnormality may be due to the presence of BCs or neurons within the dysplastic white matter or to abnormal or advanced myelination secondary to frequent seizures activity (14). In FCD without BCs, the lesions may be evident only as areas of blurring of the cortical gray–white matter junction. Cortical calcifications are occasionally demonstrated by CT. On imaging, FCD may be overt or extremely subtle and may occur in any lobe.
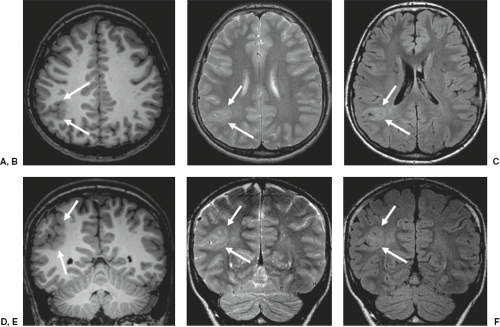 FIGURE 4.22 A 14-year-old boy with right parietal focal epileptic seizures and focal cortical dysplasia type II. Axial (A) T1-weighted, (B) T2-weighted, and (C) FLAIR images; and coronal (D) T1-weighted, (E) T2-weighted, and (F) FLAIR images show an abnormal gyral pattern with T2- and FLAIR-hyperintense and thickened cortex in the right parietal lobe (arrows). In addition, the subcortical white matter has a T1-hypointense and T2- and FLAIR-hyperintense signal. |
Bottom-of-sulcus dysplasia refers to a malformation of a form of FCD that is characterized by cortical thickening at the bottom of a sulcus, a funnel-shaped extension of the lesion toward the ventricular surface, commonly with abnormal signal intensity, and an abnormal gyral pattern related to the bottom-of-sulcus dysplasia, sometimes with a puckered appearance (15).
Presurgical localization of FCD often requires advanced MRI techniques and postprocessing. Functional studies including SPECT and FDG-PET scanning are also frequently needed to maximize the likelihood of identifying and defining the boundaries of FCD lesions. The role of new MRI techniques such as high field strength (>3 T), arterial spin labeling (ASL), susceptibility-weighted imaging (SWI), and DTI is currently evaluated (16).
Hemimegalencephaly
Definition
Hemimegalencephaly (HME) is a rare, almost always sporadic brain malformation characterized by overgrowth limited to one cerebral hemisphere (17).
Clinical Features
Children with HME typically present in infancy or already in the neonatal period. The first neurologic symptoms/findings are usually macrocephaly without signs of increased intracranial pressure; severe, early onset, and usually intractable epileptic seizures; and global developmental delay mostly leading to cognitive impairment (17,18). Contralateral hemiparesis and ipsilateral hemianopsia have also been reported. Patients with unilateral megalencephaly usually have a large head very early in life, but, possibly as a result of the intractable seizures, the head size diminishes relative to the normal curve, and eventually the patients may be normocephalic or microcephalic.
Pathogenesis and Genetics
There are three forms of HME: (1) isolated HME, which is restricted to the hypertrophic and dysplastic hemisphere; (2) associated HME, in which HME is part of neurocutaneous syndromes, particularly the epidermal nevus syndrome, but also Klippel–Trenaunay syndrome, neurofibromatosis type 1, encephalocraniocutaneous lipomatosis, Proteus syndrome, hypomelanosis of Ito, tuberous sclerosis, megalencephaly–capillary malformation, and megalencephaly–polydactylypolymicrogyria–hydrocephalus syndromes (17); and (3) total HME in which the ipsilateral cerebellar hemisphere and the brainstem are also enlarged and show abnormal foliation (17,19).
Recently, somatic mutations in three PI3 K-AKT-mTOR pathway genes have been reported in patients with HME including PIK3CA, AKT3, and MTOR. Mutations of this pathway genes cause the activation of the pathway, which is highly expressed in the developing brain during corticogenesis. In some cases, mutations are apparently limited to the
brain. As mentioned above, HME may also occur in the setting of neurocutaneous disorders and be caused by somatic de novo mutations as part of the megalencephaly-capillary malformation and megalencephaly-polydactylypolymicrogyria-hydrocephalus syndromes. In summary, HME may be caused by mutations in brain progenitor cells, but that some of these mutations occur early enough in development to be present in many tissues, affecting cells outside the brain as well. In contrast, other mutations might be limited to the brain because they occur after the embryonic separation of brain from non-brain tissue.
brain. As mentioned above, HME may also occur in the setting of neurocutaneous disorders and be caused by somatic de novo mutations as part of the megalencephaly-capillary malformation and megalencephaly-polydactylypolymicrogyria-hydrocephalus syndromes. In summary, HME may be caused by mutations in brain progenitor cells, but that some of these mutations occur early enough in development to be present in many tissues, affecting cells outside the brain as well. In contrast, other mutations might be limited to the brain because they occur after the embryonic separation of brain from non-brain tissue.
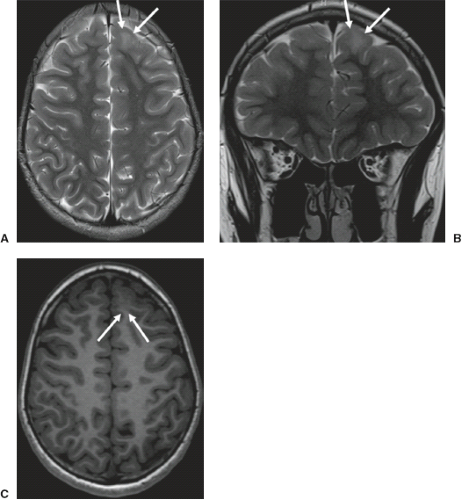 FIGURE 4.23 A 12-year-old girl with left frontal focal epileptic seizures and focal cortical dysplasia type II. (A) Axial and (B) coronal T2-weighted images show a T2-hyperintense signal of the affected cortex and underlying subcortical white matter (arrows). C: High-resolution axial T1-weighted image reveals a T1-hypointense signal of the subcortical white matter as well as an indistinct gray–white matter junction (arrows). |
Neuropathology and Imaging Findings
On neuropathology, the affected overgrown hemisphere exhibits increased white matter volume, malformations of cortical development including pachygyria, LIS, PMG, heterotopias, leptomeningeal glioneuronal heterotopia, and, most often, ipsilateral ventricular enlargement (Fig. 4.24). Malformations may be seen in the “unaffected” hemisphere as well.
Conventional neuroimaging typically shows an asymmetry between the cerebral hemispheres with moderate to marked enlargement of the affected hemisphere (Fig. 4.25) (17). The cortex of the affected hemisphere appears dysplastic with an abnormal gyral pattern including broad gyri, shallow sulci, and cortical thickening resembling LIS or pachygyria, and blurring of the cortical–white matter junction (Fig. 4.24). The affected occipital lobe may be disproportionately prominent and be displaced with the falx across the midline toward the contralateral lobe. The ipsilateral ventricle appears typically enlarged with straightening of the frontal horn and/or unilateral colpocephaly (Figs. 4.24 and 4.25). The white matter shows abnormally high T2-signal intensity due to absent or incomplete myelination (Figs. 4.24 and 4.25). Cerebral calcifications may be seen on CT. The corpus callosum is almost always asymmetric with enlargement and dysplasia of the affected side (Fig. 4.26). The ipsilateral olfactory and optic nerves may also be significantly enlarged (19). The vessels of the affected hemisphere appear also enlarged compared to the contralateral side (Fig. 4.24) (19). HME most often involves only the cerebral hemisphere; however, the brainstem and cerebellum may also be enlarged (total HME) (Fig. 4.25).
Advanced Imaging
Advanced neuroimaging, in particular DTI, may show a complete disorganization of the ipsilateral white matter fibers with an abnormal concentric orientation around the ventricle (Fig. 4.26). The septum pellucidum maybe be wide due to aberrant, midsagittal, bandlike structures beneath the corpus callosum (Fig. 4.26) connecting the frontal, parietal, and/or occipital lobes either bilaterally or ipsilaterally. Abnormal interhemispheric fibers maybe present in HME. In the majority of the cases, the ipsilateral callosal tracts are larger compared to the contralateral side because of abnormal/missing interhemispheric connections of these fibers with a resultant increased amount of ipsilateral fibers. In a minority of cases, however, the volume of the ipsilateral callosal fibers is reduced. Finally, DTI
may reveal asymmetry of other white matter tracts with thickening of the ipsilateral ones.
may reveal asymmetry of other white matter tracts with thickening of the ipsilateral ones.
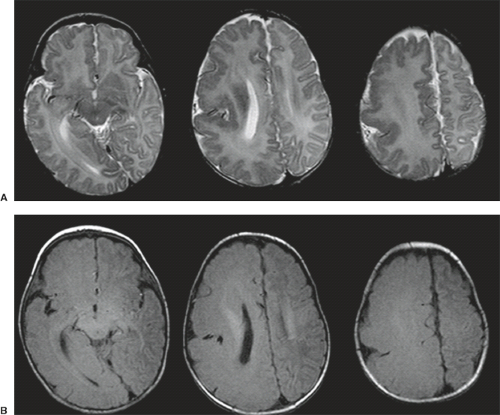 FIGURE 4.24 A 13-month-old boy with Klippel–Trenaunay–Weber syndrome and right hemimegalencephaly. Axial (A) T2-weighted and (B) T1-weighted images show an enlargement of the right cerebral hemisphere with polymicrogyria within the frontal, temporal, and parietal cortex, T1-hyperintense and T2-hypointense signals of the subcortical white matter, and dilatation of the right lateral ventricle. In addition, the ipsilesional meningeal vessels are enlarged. |
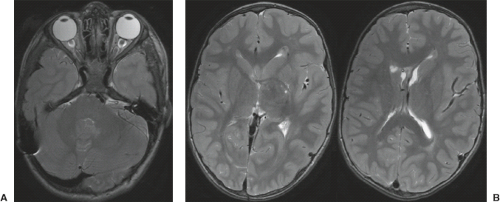 FIGURE 4.25 A 15-month-old boy with megalencephaly–capillary malformation and left hemimegalencephaly. A: Axial T2-weighted image of the cerebellum shows enlargement of the left cerebellar hemisphere (ipsilateral to the cerebral involvement). B: Axial T2-weighted images at the level of the cerebrum reveal a moderate enlargement of the left cerebral hemisphere with cortical dysplasia and T2-hyperintense signal of the left periventricular white matter. (Reprinted with permission from Poretti A, Boltshauser E. Hemicerebellar hemimegalencephaly. In: Boltshauser E, Schmahmann JD, eds. Cerebellar Disorders in Children. London: MacKeith Press; 2012.) |
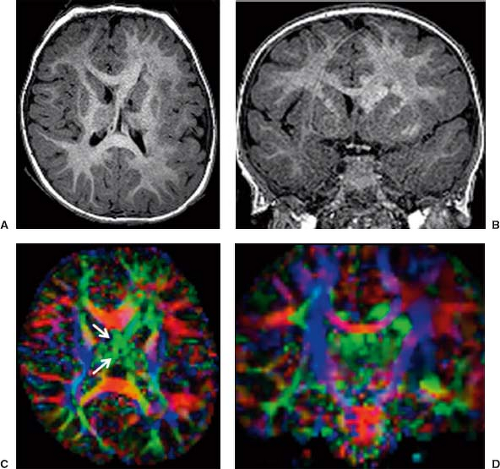 FIGURE 4.26 A 15-month-old boy with left hemimegalencephaly. A: (A) Axial and (B) coronal T1-weighted images shows enlargement of the left cerebral hemisphere. (C) Axial and (D) coronal color-coded FA maps reveal asymmetrical size of the fibers running through the anterior and posterior parts of the corpus callosum (increased size within the megalencephalic hemisphere) as well as abnormal white matter tracts within the megalencephalic hemisphere (white arrows). (Reprinted with permission from Poretti A, Meoded A, Rossi A, et al. Diffusion tensor imaging and fiber tractography in brain malformations. Pediatr Radiol 2013;43:28–54.) |
Malformations Secondary to Abnormal Neuronal Migration
Cobblestone Malformation
Definition
Cobblestone malformation is a severe brain malformation associated with abnormal migration from the brain into the leptomeninges, and frequently with eye anomalies and congenital muscular dystrophy (CMD) (20). Cobblestone malformation occurs in CMD due to reduced O-glycosylation and rarely N-glycosylation of α-dystroglycan. Based on the severity of the findings, different phenotypes have been described in order of increasing severity: Fukuyama congenital muscular dystrophy (FCMD), muscle–eye–brain disease (MEB), and Walker–Warburg syndrome (WWS).
Clinical Features
The clinical phenotype of patients with cobblestone malformation is characterized by muscle (proximal weakness, hypotonia, and increased creatine kinase values), brain (intellectual disability, seizures, tetraspasticity, and, in some patients, macrocephaly), and eye (micro-ophthalmia, optic nerve hypoplasia, chorioretinal coloboma, retinal dysplasia, cataract, glaucoma, or high myopia) involvement (20).
Genetics and Pathogenesis
To date, mutations in 15 genes have been associated with α-dystroglycanopathies. Mutations in these genes cause a reduced O-glycosylation and rarely N-glycosylation of α-dystroglycan. α-dystroglycan is a highly glycosylated peripheral membrane protein that binds many extracellular matrix proteins through attached carbohydrate groups. In α-dystroglycanopathies, glycosyl groups are absent or reduced, resulting in decreased binding of ligands such as laminin-2, agrin, and perlcan in skeletal muscles and neurexin in the brain. This causes defects in the pial limiting membrane and its attachment to radial glial fibers.
Mutations within the 15 individual genes are associated with a phenotypic spectrum. Mutations in the majority of the genes may cause different phenotypes and there is not a real phenotype–genotype correlation. Only patients with POMGnT1 mutations are somehow exceptional because the vast majority of them present with features of MEB (20).
Neuropathology and Imaging Findings
The neuroimaging findings of α-dystroglycanopathies are wide and range from severe changes of WWS to normal brain MRI (20). However, the majority of the patients fit into one of the phenotypes (FCMD, MEB, WWS, CMD with cerebellar hypoplasia/dysplasia, normal brain MRI).
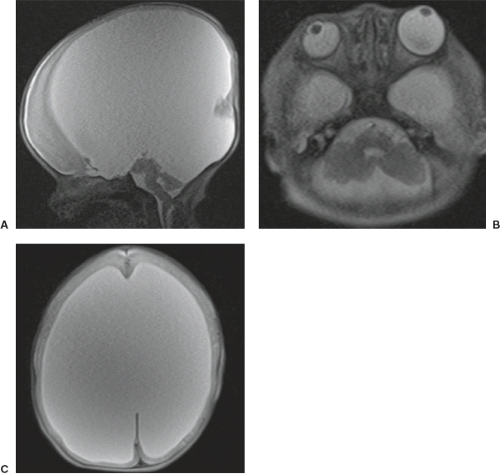 FIGURE 4.27 A 1-day-life-old neonate with Walker–Warburg syndrome and POMT2 mutations. (A) Midsagittal and (B,C) axial T2-weighted images show a marked ventriculomegaly, a diffusely thin cerebral cortex with almost no sulcations, severe hypoplasia of the cerebellar vermis and hemispheres, and an abnormal kinking of the brainstem with elongation and thickening of the midbrain and tectum and marked pontine hypoplasia. In addition, an asymmetry in the size of the eye globes with right micro-ophthalmia was seen. |
In WWS, hydrocephalus is consistently present and causes macrocephaly and prominent forehead (Fig. 4.27). The cerebral surface is undersulcated and there is usually diffuse agyria. The cerebral cortex is moderately thick, unless thinned due to hydrocephalus. The cortical–white matter junction is jagged with common vertical striations. The white matter has a very abnormal signal (T2 hyperintense and T1 hypointense). The corpus callosum is typically thin. The presence of an occipital encephaloceles is not uncommon. The brainstem and cerebellum are highly dysplastic with a pontomesencephalic kinking (21). The pons is usually hypoplastic and has a ventral midline cleft. The tectum and midbrain, however, are usually enlarged and dysplastic. The cerebellum is globally hypoplastic, but the vermis is typically more severely affected. The cerebellar foliae have a dysplastic orientation. Cerebellar cysts are uncommon in WWS.
In MEB, neuroimaging findings are usually similar to WWS, but less severe (Fig. 4.28) (20). Involvement of the cerebral cortex is characterized by pachygyria with frontal predominance instead of agyria. Areas resembling PMG are seen too. In MEB, there is no brainstem kinking, and the cerebellar cysts are present in almost all patients. Malformative cerebellar cysts are characteristic, but not specific for α-dystroglycanopathies and are located particularly in the boundary between the normal and dysplastic cerebellar cortex (22).
In FCMD, neuroimaging findings are less severe compared to MEB (20). Hydrocephalus is uncommon and the brainstem is usually normal. Cerebellar cysts are rather common.
The prenatal diagnosis of α-dystroglycanopathies is challenging. Ventriculomegaly, if present, is nonspecific. A smooth cerebral cortex and a small cerebellum may be physiologic until 24 to 25 and 18 to 20 weeks of gestation, respectively. Cerebellar cysts develop only postnatally. The abnormal brainstem angulation seems to be the most specific prenatal imaging finding and, if associated with prenatal ventriculomegaly, should raise the suspicion of α-dystroglycanopathies.
Lissencephaly and Subcortical Band Heterotopia Spectrum
Definition
LIS or smooth brain and subcortical band heterotopia (SBH) are the classic malformations associated with abnormal neuronal migration (23). LIS is characterized by absent or abnormally wide gyri and an abnormally thick cortex. SBH consists of a normal or mildly simplified gyral pattern with a smooth band of gray matter in the superficial and middle portions of the white matter.
Clinical Features
Typically, children with LIS and SBH come to medical attention during the first year of life because of seizures, poor feeding, hypotonia, and developmental delay. Seizures have multiple semiologies including infantile spasms with classic hypsarrythmia, and are usually therapy resistant. Neurologic outcome is typically related to the grade of LIS or thickness of SBH.
Pathogenesis and Genetics
LIS, SBH, and LIS with cerebellar hypoplasia are consistently malformative in origin. To date, mutations in 12 genes have been associated with LIS and SBH and account for up to 90% of patients (23). Deletions and mutations in LIS1 are the most common causes of LIS.
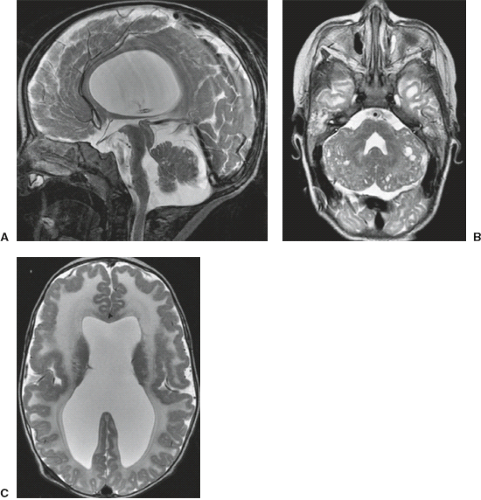 FIGURE 4.28 A 5-month-old boy with muscle-eye-brain disease and POMGnT1 mutations. (A) Midsagittal and (B,C) axial T2-weighted images show a marked ventriculomegaly, dysplastic brainstem including elongated and thickened midbrain and tectum as well as hypoplastic pons with a midline dorsal cleft, global cerebellar hypoplasia with multiple cortical/subcortical cysts within the cerebellar hemispheres and vermis, generalized polymicrogyria, diffuse T2-hyperintense signal of the supratentorial white matter, and absence of the septum pellucidum. (Reprinted with permission from Poretti A, Boltshauser E, Doherty D. Cerebellar hypoplasia: differential diagnosis and diagnostic approach. Am J Med Genet C Semin Med Genet 2014;166C:211–226.) |
The classic four-layered LIS is associated with a spectrum of malformations including isolated LIS, SBH, and Miller–Dieker syndrome. Miller–Dieker syndrome is characterized by severe four-layered LIS with agyria and not clear gradient (Fig. 4.29), characteristic facial dysmorphism (e.g., prominent forehead, short nose with upturned nares, and protuberant upper lip), and additional body malformations. The genotype of Miller–Dieker syndrome consists of large deletions in chromosome 17p13.3 that include LIS1, YWHAE, and all intervening genes.
Isolated LIS consists of classic four-layered LIS with normal or mildly hypoplastic cerebellum. Various genes have been associated with different patterns. Boys with DCX mutations have a diffuse severe agyria or a frontal predominant LIS. Children with TUBA1 A mutations have also a posterior predominant LIS.
SBH is caused by heterozygous mutations in DCX and most patients are female.
LIS with cerebellar hypoplasia maybe divided into two groups. The first group consists of mild frontal predominant LIS associated with severe hippocampal and cerebellar hypoplasia and dysplasia. Mutations in RELN (encodes an extracellular matrix-associated glycoprotein (reelin) that is secreted by Cajal–Retzius cells in the developing cerebral cortex and is critical for the regulation of neuronal migration during cortical and cerebellar development) and VLDLR (an essential cell-surface receptor for reelin) have been found in this group of LIS patients (24,25). The second group consists of more severe LIS with cerebellar hypoplasia as occurs in tubulinopathies (26).
Neuropathology and Neuroimaging Findings
In all LIS forms, the brain surface appears smooth with areas of absent (agyria: the sylvian fissures are the only definable fissures) or abnormally wide (pachygyria) gyri, and vertically oriented sylvian fissures. The cerebral cortex is thickened (8 to 15 mm compared to 2.5 to 4 mm of the normal cortex). In SBH, the brain surface is normal and there is a smooth band of misplaced gray matter within the subcortical white matter (Fig. 4.30).
The anterior to posterior gradient, gender distribution, and associated malformations are essential for recognition of the different genetic forms. Mutations of DCX, ACTB, and ACTG1 result in an anterior–posterior gradient, while mutations of LIS1, TUBA1 A, TUBG1, and DYNC1H1 have a posterior–anterior gradient (23) (Fig. 4.31). Severe LIS with agenesis of the corpus callosum in a boy is strongly suggestive of ARX mutations (Fig. 4.32). LIS with pontocerebellar hypoplasia (PCH) suggests RELN or VLDRL mutations. The vermis is typically more affected compared to the hemispheres and typically does not show almost any foliation. Pontine hypoplasia is very consistent. Neuroimaging findings are more severe in RELN mutation compared to VLDRL mutation.
Neuronal Heterotopia
Definition
Neuronal heterotopia consists of a group of neurons in an inappropriate location. The major types include PNH that line the lateral ventricles, subcortical nodular heterotopia, that tend to form large masses of nodules beneath the cortex, and SBH. SBH is discussed with LIS because of the similar pathomechanism.
Periventicular Heterotopia
PNH is the most common form of neuronal heterotopia. Anatomically, PNH
consists of nodular masses of gray matter that line the ventricular walls and protrude into the lumen, resulting in an irregular outline.
consists of nodular masses of gray matter that line the ventricular walls and protrude into the lumen, resulting in an irregular outline.
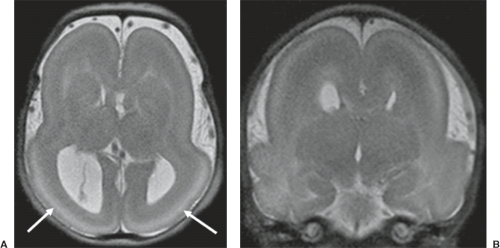 FIGURE 4.29 A 1-day-old neonate with Miller-Dieker syndrome and lissencephaly. (A) Axial and (B) coronal T2-weighted images show absence of medial hemispheric sulci other than the parieto-occipital sulcus and a smooth cerebral surface with the typical cortical pattern with thin (dark) outer cortical layer, relatively thick (bright) cell-sparse zone (arrows), and thick (dark) inner cortical layer. The lateral ventricles are relatively enlarged. |
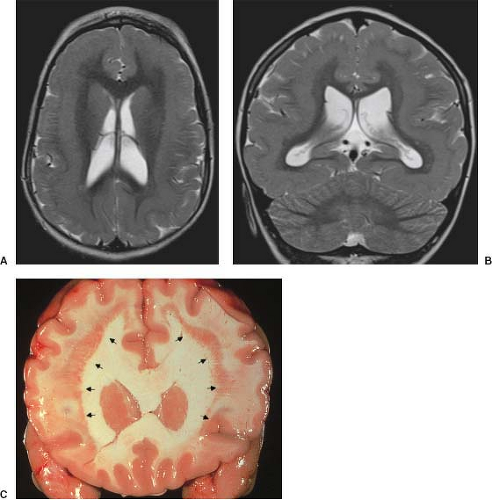 FIGURE 4.30 A 7-year-old girl with intractable seizures, developmental delay, and subcortical band heterotopia due to DCX mutations. (A) Axial and (B) coronal T2-weighted images show the gray–matter intensity band separated from the cortex by a layer of partially myelinated white matter. In addition, the cortex has normal thickness, but shallow sulci, and the lateral ventricles are mildly enlarged. C: Coronal necropsy specimen shows band of heterotropic gray (arrows) between subcortical and deep white matter. |
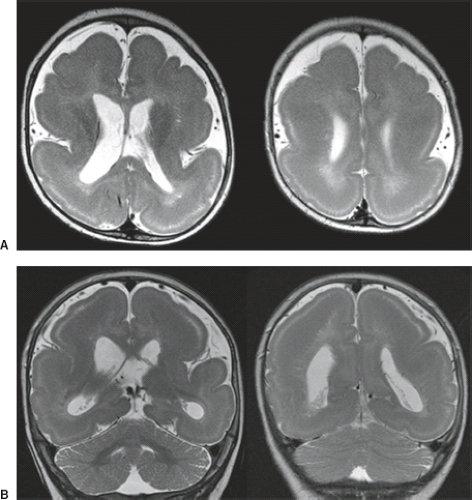 FIGURE 4.31 A 4-month-old boy with intractable seizures, developmental delay, and lissencephaly due to LIS1 mutations. (A) Axial and (B) coronal T2-weighted images show pachygyria in the posterior lobes and a more normal gyral pattern anteriorly (posterior to anterior gradient). The posterior fossa contents are unremarkable. |
Seizures and learning problems are common, whereas more severe developmental problems are uncommon. The age at seizure onset is variable. Most patients have focal seizures, which can not be easily controlled or are refractory to treatment.
The most common form of diffuse PNH is caused by mutations in the X-linked FLNA gene (27). Autosomal recessive mutations in ARFGEF2 have been reported in patients with
severe congenital microcephaly and diffuse bilateral PNH. FLNA and ARFGEF2 regulate actin binding, vesicle trafficking, cell adhesion, and function of radial glia.
severe congenital microcephaly and diffuse bilateral PNH. FLNA and ARFGEF2 regulate actin binding, vesicle trafficking, cell adhesion, and function of radial glia.
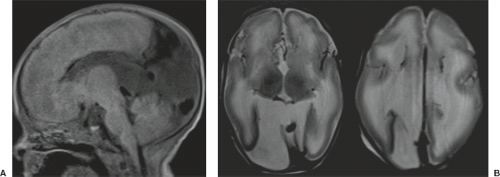 FIGURE 4.32 A 5-day-old male neonate with epileptic seizures, ambiguous genitalia, and lissencephaly due to ARX mutation. A: Sagittal T1-weighted image shows absence of the corpus callosum and abnormal sulcation of the medial cerebral hemisphere. B: Axial T2-weighted images reveal area of pachygyria and agyria with moderately thick cortex and almost absence of the basal ganglia. |
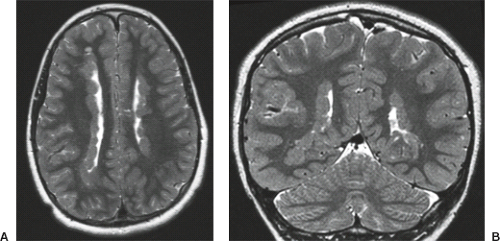 FIGURE 4.33 A 7-year-old boy with complex partial seizures and bilateral periventricular nodular heterotopia due to FLNA mutation. (A) Axial and (B) coronal T2-weighted images show bilateral diffuse nodular heterotopia along the wall of the lateral ventricles. |
On MRI, PNH appears as ovoid lesions within the subependymal region. They are isointense with gray matter on all imaging sequences (Figs. 4.33 and 4.34). Neither perilesional edema nor contrast enhancement is seen. PNH may include only one or two lesions or may include nodules along the length of the lateral ventricles. Patients with FLNA mutations typically have bilateral contiguous PNH that spares the temporal horns, and mild cerebellar vermis hypoplasia with mega cisterna magna (Fig. 4.33) (27). Patients with ARFGEF2 mutations and PNH can have severe congenital microcephaly and thin overlying cortex with abnormal gyri. PNH may be limited to the trigons, temporal, and occipital horns of the lateral ventricles and can be associated with overlying PMG, hippocampal and cerebellar hypoplasia, or hydrocephalus.
The differential diagnosis of subependymal heterotopia is limited and includes tuberous sclerosis and ependymal metastases, especially from medulloblastoma in the pediatric population. In general, the lesions of tuberous sclerosis are iso- to hypointense to white matter, and they may or may not enhance. In addition, other manifestations of tuberous sclerosis are often present. In patients with ependymal metastases, a history of the primary tumor is usually available, and enhancement is the rule, although not without exception. In cases of medulloblastoma metastases to the ependyma of the lateral and third ventricles, enhancement may be lacking.
Subcortical Nodular Heterotopia
Subcortical nodular heterotopia consists of a large mass of nodules expanding a portion of one cerebral hemisphere. Subcortical nodular heterotopias occur at any location within the subcortical white matter and may be associated with ipsilateral PNH, agenesis of the corpus callosum, and hypoplasia of the cerebellar vermis (28).
Malformations Secondary to Abnormal Cortical Organization and Late Migration
Polymicrogyria With or Without Schizencephaly
Definition
The term PMG describes a cerebral cortex with many excessively small convolutions, which might or might not be visible on gross inspection of the brain surface (29,30). One specific pattern of PMG occurs in schizencephaly. The presence of PMG along the cleft is part of the definition of schizencephaly. The pathogenesis of PMG is poorly understood and is most likely heterogeneous (31).
Clinical Features
Patients with PMG may have a variable clinical presentation, which depends on several factors. The occurrence of severe microcephaly, abnormal neurologic examination (particularly spasticity), widespread distribution of
PMG, and additional brain malformations (especially cerebellar hypoplasia) are predictors of poor outcome, while patients with focal unilateral PMG have the best outcome (Fig. 4.35). Depending on the location, patients with unilateral PMG may present with mild hemiparesis. In addition, they may develop seizures. Bilateral PMG within the perisylvian region may cause oromotor dysfunction (suprabulbar palsy), seizures, and intellectual disability (Fig. 4.36). In patients with PMG and schizencephaly, the presence of open-lip schizencephaly is a predictor of poorer outcome (Fig. 4.37). Patients with PMG and closed-lip schizencephaly usually present with hemiparesis or motor delay (Fig. 4.38), while patients with PMG and open-lip schizencephaly usually present with hydrocephalus and seizures.
PMG, and additional brain malformations (especially cerebellar hypoplasia) are predictors of poor outcome, while patients with focal unilateral PMG have the best outcome (Fig. 4.35). Depending on the location, patients with unilateral PMG may present with mild hemiparesis. In addition, they may develop seizures. Bilateral PMG within the perisylvian region may cause oromotor dysfunction (suprabulbar palsy), seizures, and intellectual disability (Fig. 4.36). In patients with PMG and schizencephaly, the presence of open-lip schizencephaly is a predictor of poorer outcome (Fig. 4.37). Patients with PMG and closed-lip schizencephaly usually present with hemiparesis or motor delay (Fig. 4.38), while patients with PMG and open-lip schizencephaly usually present with hydrocephalus and seizures.
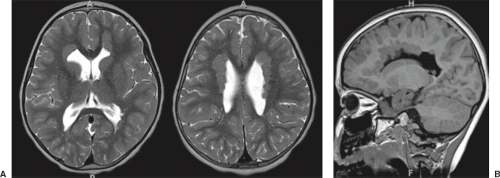 FIGURE 4.34 A 3-year-old girl with complex partial seizures and bilateral periventricular nodular heterotopia. (A) Axial T2-weighted and (B) sagittal T1-weighted images reveal bilateral nodular heterotopia along the anterior wall of the lateral ventricles and extending into the white matter lateral to the ventricular bodies. |
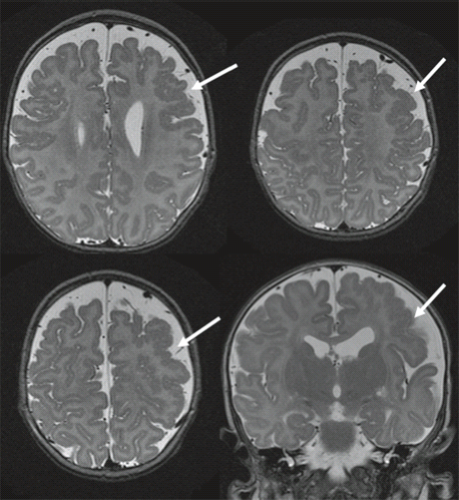 FIGURE 4.35 Focal polymicrogyria. A 3-year-old girl with complex partial seizures. Axial and coronal T2-weighted images show irregularity of the individual gyri over the left frontal lobe representing polymicrogyria (arrows). |
Genetics and Pathogenesis
The pathogenesis of PMG is heterogeneous and includes malformative and acquired
causes. Acquired causes include congenital infections (particularly cytomegalovirus, Fig. 4.39) and vascular insufficiency (especially during twin pregnancies) (32). Mutations in several genes with all types of inheritance (e.g., SRPX2, RAB3GAP1, EOMES, TUBB2B, COL18A1, KIAA1279, GPR56, and PAX6) have been associated with PMG (23,31). In addition, some copy-number variants have been associated with PMG, but only deletions in 1p36.3 and 22q11.2 are common. PMG may be part of well-defined syndrome such as Aicardi syndrome, oculocerebrocutaneous syndrome, DiGeorge syndrome, and Warburg Micro syndrome. Finally, PMG has been reported in metabolic disorders such as Zellweger syndrome (Fig. 4.40), neonatal adrenoleukodystrophy, and glutaric aciduria type 2, although the histopathology differs from classic PMG.
causes. Acquired causes include congenital infections (particularly cytomegalovirus, Fig. 4.39) and vascular insufficiency (especially during twin pregnancies) (32). Mutations in several genes with all types of inheritance (e.g., SRPX2, RAB3GAP1, EOMES, TUBB2B, COL18A1, KIAA1279, GPR56, and PAX6) have been associated with PMG (23,31). In addition, some copy-number variants have been associated with PMG, but only deletions in 1p36.3 and 22q11.2 are common. PMG may be part of well-defined syndrome such as Aicardi syndrome, oculocerebrocutaneous syndrome, DiGeorge syndrome, and Warburg Micro syndrome. Finally, PMG has been reported in metabolic disorders such as Zellweger syndrome (Fig. 4.40), neonatal adrenoleukodystrophy, and glutaric aciduria type 2, although the histopathology differs from classic PMG.
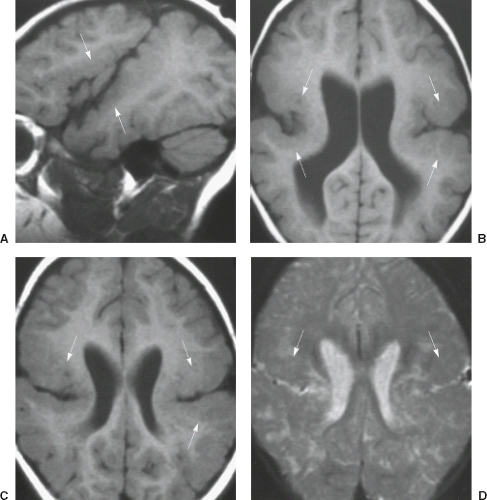 FIGURE 4.36 Bilateral perisylvian syndrome. (A) Sagittal and (B,C) axial T1-weighted, and (D) axial T2-weighted images show bilateral anomalies of cortical development (arrows) overlying underdeveloped sylvian fissures. Note the dorsal, perirolandic extension of the sylvian fissures, typical of anomalies of perisylvian polymicrogyria. Bodies of lateral ventricles (C,D) show an inverted appearance typical of this disorder. (From Truwit C, Lempert T. Pediatric neuroimaging: a casebook approach. Denver, CO: DPS Press, 1991, with permission.) |
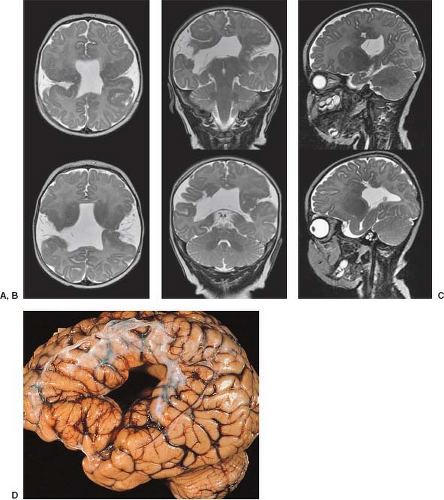 FIGURE 4.37 Bilateral open-lip schizencephaly. (A) Axial, (B) coronal, and (C) sagittal T2-weighted images show bilateral gray matter–lined clefts and absence of the septum pellucidum. The gray matter lining of the clefts is thick and irregular, suggesting polymicrogyria. The CSF is continuous from the subarachnoid space to the ventricle (open-lip). D: Lateral view of a necropsy brain specimen from a patient with a lifelong history of seizures shows a transcerebral schizencephalic cleft communicating with the lateral ventricle and dysmorphic frontal gyri. |
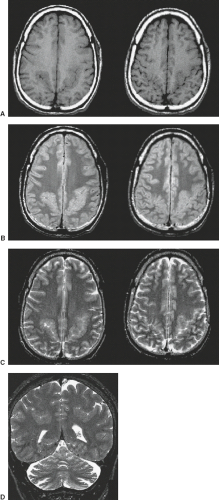 FIGURE 4.38 Bilateral closed-lip schizencephaly. (A) Axial T1-weighted, (B) axial proton density–weighted, (C) axial T2-weighted, and (D) coronal T2-weighted images show bilateral thickened polymicrogyric cortex of the parietal lobes without communication between the clefts and lateral ventricles. Mild global volume loss of the cerebellum with widened fissures. |
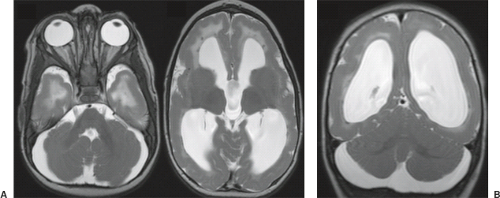 FIGURE 4.39 A 14-month-old child with confirmed congenital cytomegalovirus infection. (A) Axial and (B) coronal T2-weighted images show cerebellar hypoplasia, enlarged lateral ventricles, diffuse T2-hyperintense signal of the cerebral white matter, and diffuse cortical gyral anomaly resembling polymicrogyria. |
Schizencephaly has been linked to EMX2 mutations, but this finding has never been confirmed. Recently, mutations in COL4A1 have been associated with schizencephaly (6). Dominant mutations in COL4A1 cause changes of the basal membrane of capillaries resulting in microangiopathy and, hence, prenatal hemorrhages and/or ischemias resulting in schizencephaly.
Neuropathology and Imaging Findings
In PMG, the cerebral cortex can have multiple small, delicate gyri or appear thick and irregularly bumpy or be paradoxically smooth because the outer cortical (molecular) layer fuses over the microsulci (Fig. 4.41). Because of the immature myelination, in young children with PMG the cortex may not appear particularly thickened. Irregularities of the gray–white matter junction are often the most convincing evidence of PMG (Fig. 4.35) (29). PMG may occur in several locations (33). Involvement of the bilateral perisylvian region is the most common location (Fig. 4.36) and may vary from the posterior perisylvian region only, to the entire perisylvian region, to the perisylvian region with extension to other brain regions but not the poles, to most of the brain including either or both the frontal or occipital poles. Perisylvian PMG can be bilateral symmetrical, bilateral asymmetrical, or unilateral. In bilateral perisylvian PMG, the opercula are dysplastic and incomplete and the sylvian fissure is wide and underdeveloped. Sagittal images may show posterior extension of the sylvian fissure, exposure of the insula, and apparent thickening of the cortex. Other patterns are almost always bilateral and symmetrical, and include generalized, frontal, posterior (probably), mesial parieto-occipital, and rare diffuse parasagittal PMG (29,33). PMG may be associated with other malformations including corpus callosum agenesis and hypogenesis, cerebellar hypoplasia, PNH, and subcortical
heterotopia (29). PMG may be associated with anomalous venous drainage and large vessels are especially common in regions where there is a large infolding of thickened cortex. In PMG, diffusely abnormal signal in white matter should suggest prenatal infection, a peroxisomal disorder or a PMG-like disorder such as cobblestone brain or GPR56 mutation.
heterotopia (29). PMG may be associated with anomalous venous drainage and large vessels are especially common in regions where there is a large infolding of thickened cortex. In PMG, diffusely abnormal signal in white matter should suggest prenatal infection, a peroxisomal disorder or a PMG-like disorder such as cobblestone brain or GPR56 mutation.
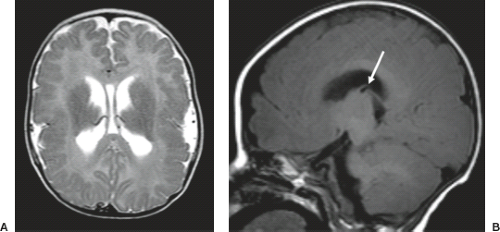 FIGURE 4.40 Newborn with confirmed Zellweger syndrome. (A) Axial T2-weighted and (B) sagittal T1-weighted images show diffuse predominantly perisylvian polymicrogyria and a subependymal cyst just anterior to the caudate vein (arrow) in the caudothalamic groove. |
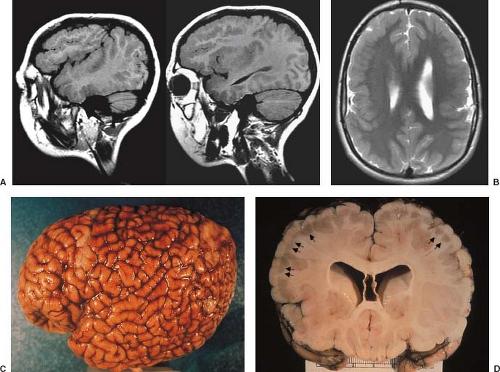 FIGURE 4.41 Polymicrogyria. A: Sagittal T1-weighted and (B) axial T2-weighted images show a markedly thickened cortex bilaterally with diagnostic irregularity at the interface of gray matter and subcortical white matter. C: The surface morphology of innumerable, abnormally small gyral contours indicative of polymicrogyria. On section (D), polymicrogyria is seen as areas of gray matter forming “pseudogyri” (arrows) that lack true sulci and therefore present as thick, nearly smooth cortical surfaces. |
In schizencephaly, the gray matter lining the cleft has the imaging appearance of PMG with an irregular surface, deep infolding (the cleft), mildly thick cortex, and stippling of the interface between gray and white matter (Fig. 4.42). Schizencephaly is often bilateral but frequently asymmetrical; the contralateral hemisphere should be closely assessed for milder clefts or PMG without cleft. In bilateral schizencephaly, the septum pellucidum is typically missing (Fig. 4.37).
Fetal MRI may show the schizencephalic cleft. Prenatally, most clefts are open. However, up to the half of them was found to have subsequently closed on postnatal MRI (34).
Tubulinopathies
Definition
Tubulinopathies is a recently described group of brain malformation that is caused by mutations in genes that function during the early stages of neuronal proliferation, migration, differentiation, and axonal guidance (26). The full phenotypic spectrum of tubulinopathies is not yet fully known, but is vary from severe LIS with cerebellar hypoplasia to less severe malformation such as PMG.
Clinical Features
Severe intellectual disability and intractable seizures are the most common features of tubulinopathies. Tetraspastic cerebral palsy and postnatal microcephaly are other neurologic findings. Dysmorphic features are rare and other organs are not affected.
Genetics and Pathogenesis
To date, mutations in nine genes (DYNC1H1, KIF2A, KIF5C, TUBA1A, TUBA8, TUBB, TUBB2B, TUBB3, and TUBG1) have been associated with tubulinopathies. The majority of mutations in tubulin genes are sporadic and de novo, but germline mosaicism and autosomal recessive inheritance have also been observed. The tubulinopathies genes play a role in the regulation of microtubule-dependent mitotic processes in progenitor cells, and on the trafficking activities of the microtubule-dependent molecular motors KIF2A, KIF5C, and DYNC1H1 in postmitotic neuronal cells. Some degree of phenotype–genotype correlation has been shown (26). A severe LIS with agenesis of the corpus callosum and severe PCH has been associated with mutations in TUBA1A and TUBB2B.
Imaging Findings
The spectrum of neuroimaging findings is variable and range from severe LIS with completely absent gyri, total agenesis of the corpus callosum, and severe cerebellar hypoplasia to a PMG-like malformation with cerebellar hypoplasia (26). In less severe LIS forms, a posterior to anterior gradient is usually seen. A dysmorphic appearance of the
basal ganglia (mostly putamen and caudate) with absence of the anterior limb of the internal capsule is the most characteristic and consistent finding (Fig. 4.43) (26,35). Ventriculomegaly with abnormal shape of the frontal horns, as well as agenesis/dysgenesis of the corpus callosum and anterior commissure have also been described. Posterior fossa involvement includes different degrees of PCH, cerebellar (diagonal folia across vermis in axial view) and tectal dysplasia, and asymmetric midbrain and pons.
basal ganglia (mostly putamen and caudate) with absence of the anterior limb of the internal capsule is the most characteristic and consistent finding (Fig. 4.43) (26,35). Ventriculomegaly with abnormal shape of the frontal horns, as well as agenesis/dysgenesis of the corpus callosum and anterior commissure have also been described. Posterior fossa involvement includes different degrees of PCH, cerebellar (diagonal folia across vermis in axial view) and tectal dysplasia, and asymmetric midbrain and pons.
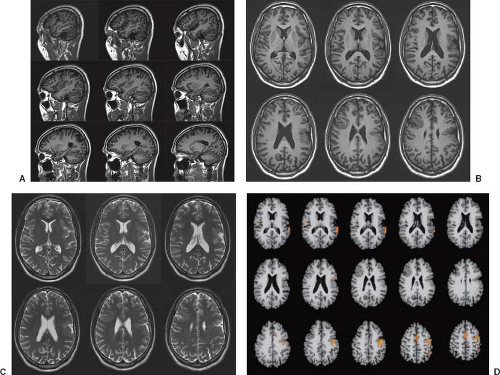 FIGURE 4.42 Schizencephaly. A: Sagittal T1-weighted images show extensive polymicrogyria with a cleft extending toward the dimpled left lateral ventricle. Axial (B) T1-weighted and (C) T2-weighted images confirm bilateral polymicrogyria, more extensive on the left side, and schizencephalic cleft. D: Preoperative functional magnetic resonance imaging in a patient with refractory epilepsy shows activity associated with motor function in proximity to abnormal cortices. |
Advanced Imaging
In tubulinopathies, DTI and tractography studies showed several white matter brain malformations, raising the intriguing hypothesis that mutations in the tubulin genes superfamily cause primary generalized defects in axon guidance (36). As consistent features, in these patients DTI and tractography reveal pontine abnormalities (abnormal course of transverse pontine fibers, Fig. 4.43D), defects in commissural fiber tracts (anterior commissure and corpus callosum abnormalities/agenesis), and within the fornix confirming the key role of tubulin genes in axon tract formation.
Anomalies of Ventral Prosencephalon Development
The disorders that cause anomalies of ventral prosencephalon development may be divided into those that involve underexpression of ventralizing gradient genes such as HPE, those that involve overexpression of dorsalizing gradient genes such as the interhemispheric variant of HPE, and those that involve underexpression of dorsalizing gradient genes such as septo-optic dysplasia (SOD). It is of note that although traditionally SOD has been considered a mild form of HPE, according to Sarnat, SOD and the interhemispheric variant of HPE are classified separately from the other types of HPE (2).
Holoprosencephaly
Definition
HPE is a complex brain malformation characterized by an incomplete cleavage of the prosencephalon (telencephalon and diencephalon) and concomitant absence or poor development of the associated midline structures like, for example, the corpus callosum, pituitary gland, and hippocampal commissure (37,38). This spectrum of malformation involves the most rostral end of the neural tube and the premaxillary segment of the face. Conceptually, the most rostral midline sections of the brain and face are not genetically induced, and, therefore, do not develop. The severity of brain and facial deformities varies widely in HPE. The clinical manifestations, in terms of normal development and neurologic function, vary with the amount of brain dysgenesis. Therefore, DeMyer divided HPE into three subcategories: alobar, semilobar, and lobar HPE. These categories are useful for classifying HPE of different severity. It should
be noted, however, that there is no clear distinction among these different categories; in reality, they represent a spectrum of brain dysplasia, ranging from the most severe form of alobar to the more nearly normal forms of lobar HPE.
be noted, however, that there is no clear distinction among these different categories; in reality, they represent a spectrum of brain dysplasia, ranging from the most severe form of alobar to the more nearly normal forms of lobar HPE.
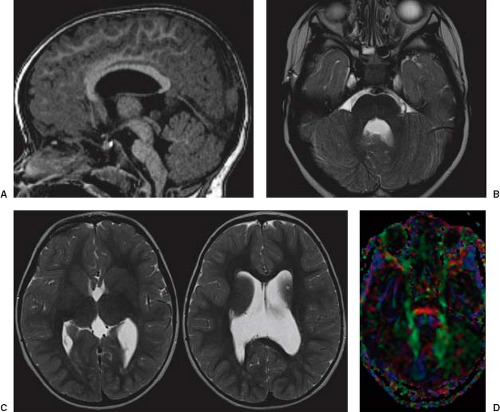 FIGURE 4.43 A 23-month-old child with developmental delay and TUBB3 mutation. A: Sagittal T1-weighted image shows cerebellar vermis hypoplasia, elongated midbrain, short/small pons, loss of the normal flat dorsal surface of the brainstem, and absence of the anterior commissure. B: Axial T2-weighted image though the cerebellum shows diagonal folia across the vermis. C,D: Axial T2-weighted images reveal dysmorphic caudate heads and putamina with loss of a clear interface between these structures and the internal capsule, bilateral ventriculomegaly with abnormal configuration of the anterior horns of the lateral ventricles, and no apparent cortical migrational abnormality. D: Axial color-coded FA maps shows absence of the ventral part of the transverse pontine fibers. |
Clinical Features
The prevalence of HPE is believed to be approximately 1 in 250 during embryogenesis, with a live birth prevalence of 1 in 10,000 to 1 in 20,000. The female-to-male ratio is 2:1, with a rate of prevalence 4.2 times higher in infants of mothers younger than 18 years of age. The alobar form is most common (46% to 54%), followed by the semilobar (18%), lobar (10%), and unclassifiable subtypes (18%).
Approximately 10% of cases have isolated intracranial abnormalities without other craniofacial defect. Craniofacial abnormalities are common and range from severe (cyclopia) to mild (single midline maxillary incisor). Eye malformations appear to be the most frequently associated (77%), followed by malformations of the nose (70%), ear (50%), and maxilla (oral clefts) (42%). Although the often-quoted remark of DeMyer that “the face predicts the brain” appears to be valid in about 80% of cases, but there are exceptions. For instance, alobar HPE may occur without craniofacial abnormality.
A high rate of mortality has been reported, with survival at 1 year of 54% for isolated HPE, 14% for syndromic HPE, and 25% for nonsyndromic HPE with multiple defects. Among infants with isolated HPE, those with the alobar form are most severely affected, with 1-year survival reported to be 20% to 30%. For those with isolated semilobar and lobar HPE, survival well into adulthood is not uncommon.
Clinical symptoms include cognitive impairment, developmental delay, hyposmia, seizures, spasticity with poor muscle control, movement disorders, pituitary dysfunction/endocrinopathies, emotional lability with sudden mood swings, hoarse or “barking” voice, growth delay, and brainstem dysfunction, including respiratory difficulties, dysrhythmias, dysphagia, and fluctuations in temperature.
Pathogenesis and Genetics
HPE is etiologically heterogeneous, and there appear to be both environmental and genetic causes. The strongest teratogenic evidence exists for maternal insulin-dependent diabetes mellitus as well as exposure to alcohol and retinoic acid. In addition, veratrum californicum, a plant that contains cyclopamine, a steroidal alkaloid, is believed to interfere with the cholesterol synthesis and with the molecular signaling along the so-called SHH gene pathway, which plays a key role in the pathogenesis of HPE
(see below). Finally, prenatal infections such as cytomegalovirus, toxoplasma, and rubella have also been linked to the occurrence of HPE in rare cases (37).
(see below). Finally, prenatal infections such as cytomegalovirus, toxoplasma, and rubella have also been linked to the occurrence of HPE in rare cases (37).
HPE is genotypically heterogeneous. Chromosomal abnormalities may be present in up to 30% to 40% of patients. HPE has a higher prevalence in trisomy 13 and 18 and triploidy. In addition, HPE may be part of Mendelian disorders with a normal karyotype such as Smith–Lemli–Opitz, Pallister–Hall, and velocardiofacial syndromes. About 25% of nonsyndromic, nonchromosomal HPE cases have been associated with mutations in at least nine genes including SHH, ZIC2, SIX3, TGIF, PATCHED-1, GLI2, DISP1, NODAL, and FOXH1 (the first four are the most prevalent). Mutations in these genes are inherited mostly with an autosomal-dominant pattern and have a penetrance of 82% to 88%. However, the majority of cases are sporadic. Despite the impressive advances in the understanding of the genetics of HPE, mutations in the known HPE genes account for less than 5% of all sporadic cases of HPE.
The putative role of each of the HPE genes in brain morphogenesis is beyond the scope of this chapter. SHH provides an example of these roles, however, in that it encodes a morphogen involved in the development of the ventral neural tube. The ventral neural tube is induced from the neuroectoderm by the notochord, along the trunk region as far rostral as the mesencephalon. This notochordal–ventral neural tube induction is mediated by the morphogen produced by SHH. SHH is in fact first expressed by the notochord, which induces the neural plate to become the so-called floor plate of the neural tube. The floor plate, once formed, also expresses SHH. SHH is also believed to be involved in craniofacial development. A defect in this mechanism of induction, therefore, is believed to be at the origin of the entire subsequent cascade of events leading to the HPE phenotype. Cholesterol modification of the SHH protein, moreover, is believed to be important in the spatial restriction of SHH protein activity, and this interaction with cholesterol helps to explain the association of HPE with disorders of cholesterol metabolism such as those caused by exposure to veratrum californicum or the Smith–Lemli–Opitz syndrome, where there is an enzymatic block in cholesterol synthesis. One upshot of the molecular genetics of HPE is thus a modification of the traditional view of HPE as a failure of telencephalic and diencephalic cleavage in favor of the view of HPE as a global field defect in forebrain patterning involving the prechordal mesoderm and endoderm believed to occur within the first 4 weeks of gestation.
Neuropathology
The evolution of the terminology used in HPE epitomizes the refinement in understanding of the neuropathology. HPE was initially termed arrhinencephaly, considering the absence of the olfactory bulbs and tracts as the hallmark feature of the disorder. The term holotelencephaly was later proposed to emphasize the involvement of the entire telencephalon. Finally, the term holoprosencephaly was suggested to summarize the involvement of the diencephalon as well as the telencephalon.
The major neuropathologic and imaging findings of the various types of HPE are detailed in what follows. Certain unifying features should be kept in mind. With regard to the cerebrum, the types of HPE exhibit progressive degrees of separation of a holosphere, and with regard to the ventricles, progressive degrees of separation of a holoventricle. The olfactory bulbs and tracts are almost always absent. The hypothalamus, neurohypophysis, and adenohypophysis are usually hypoplastic and hypofunctional. The mammillary bodies may be fused. The cytoarchitecture of the cerebral cortex is a subject of debate. Although malformations of cortical development may be present, primary defects of cell migration are believed to be uncommon. Of interest, the cerebellum may show cortical dysplasia or heterotopia, particularly in cases of chromosomal HPE. There is usually an abnormal course of the middle and anterior cerebral arteries (ACAs) (Fig. 4.44).
 FIGURE 4.44 Axial MIP of a TOF MRA in a child with semilobar holoprosencephaly shows an azygous anterior cerebral artery. |
Alobar HPE: Pathology and Imaging Findings
Alobar HPE is the most severe form of HPE and is frequently associated with severe midline facial deformities resulting from absence or hypoplasia of the premaxillary segment of the face. These craniofacial abnormalities include cyclopia with fused orbits, single eyeball, fused or absent metopic suture, and forehead proboscis and cebocephaly, in which two severely hypoteloric orbits are present with a proboscis between or below the orbits.
Examination of the brain reveals complete fusion of the two cerebral hemispheres such that the cerebrum is composed of a single flattened mass of tissue that usually sits adjacent to the most rostral portion of the calvarium (Fig. 4.45) (38). There is absence of the interhemispheric fissure and falx cerebri. The corpus callosum and anterior commissure are usually absent; a rudimentary callosal plate may be present. There is a large, crescent-shaped holoventricle; the septum pellucidum is absent. The basal ganglia and thalami vary from distinct right and left hemispheric structures to complete fusion. The basal ganglia are sometimes absent. There is most often a large dorsal cyst which may communicate with the monoventrile. This cyst usually occupies more than half of the volume of the calvarium.
Semilobar HPE: Pathology and Imaging Findings
Semilobar HPE is a less severe anomaly than alobar HPE with lack of separation of the anterior part of the hemispheres. Patients with semilobar HPE usually have normal facies, but they occasionally have mild facial anomalies including clefts of the lip and palate. The interhemispheric fissure and falx cerebri are usually formed posteriorly, but are absent anteriorly. As a result, there is separation of the cerebral hemispheres posteriorly, but the frontal lobes are undivided across the midline (Fig. 4.46) (38). The hippocampal formations remain rudimentary and, as a result, the temporal horns of the lateral ventricles are large and incompletely formed. On sagittal images, an interhemispheric commissure that strongly resembles the posterior portions of the corpus callosum is often seen dorsal and superior to the trigones of the lateral ventricles. The anterior portions of the corpus callosum are always absent. It is debated in the developmental literature whether the “pseudosplenium” is really a corpus callosum. In HPE, it is a common thought that the normal sequence of formation of
the corpus callosum does not hold true. Nowadays, we know that the corpus callosum does not develop from the front to the back, but the different components develop independently and fuse at a later embryologic stage (7). In semilobar HPE, the presence of the splenium of the corpus callosum in the absence of a genu, body, and rostrum does not imply destruction of the more anterior portions of the corpus callosum, but an abnormal (malformative) development. The septum pellucidum is always completely absent. The deep gray matter nuclei are frequently incompletely separated; the thalami are, however, usually partially separated, resulting in a small third ventricle. A dorsal cyst may or may not be present. In addition to midline anomalies, semilobar HPE is commonly associated with anomalies of cortical development and disordered neuronal migration.
the corpus callosum does not hold true. Nowadays, we know that the corpus callosum does not develop from the front to the back, but the different components develop independently and fuse at a later embryologic stage (7). In semilobar HPE, the presence of the splenium of the corpus callosum in the absence of a genu, body, and rostrum does not imply destruction of the more anterior portions of the corpus callosum, but an abnormal (malformative) development. The septum pellucidum is always completely absent. The deep gray matter nuclei are frequently incompletely separated; the thalami are, however, usually partially separated, resulting in a small third ventricle. A dorsal cyst may or may not be present. In addition to midline anomalies, semilobar HPE is commonly associated with anomalies of cortical development and disordered neuronal migration.
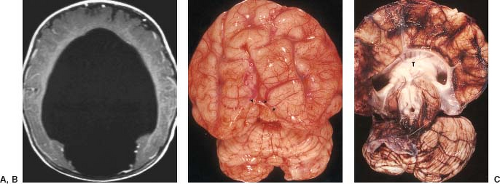 FIGURE 4.45 Alobar holoprosencephaly. A: An axial T1-weighted image shows no definable interhemispheric fissure. The falx cerebri is absent. The cerebrum is composed of a pancakelike mass of tissue situated in the rostral calvarium. A crescent-shaped holoventricle is continuous with a large dorsal cyst. B: A whole-brain necropsy specimen viewed from above shows a shallow, underdeveloped interhemispheric fissure posteriorly (arrows) and an absent interhemispheric fissure anteriorly. Note the abnormal gyral pattern, with broad and smooth gyral morphology suggestive of pachygyria. C: An axial view of a fixed specimen demonstrates monoventricle and fused thalami (T) with anterior continuation of gray and white matter and no interhemispheric fissure. Pachygyria is also present. (B,C: Courtesy of Dr. Mauricio Castillo.) |
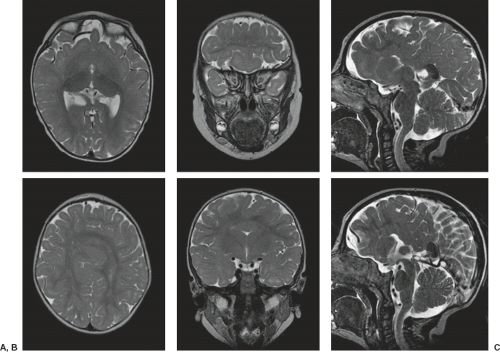 FIGURE 4.46 Semilobar holoprosencephaly. (A) Axial, (B) coronal, and (C) sagittal T2-weighted images show the fusion of the frontal lobes across the midline with an abnormal gyral pattern and global hypoplasia of the frontal lobes, incomplete separation of the thalami, presence only of the splenium of the corpus callosum, and absence of the anterior horns of the lateral ventricles. |
Lobar HPE: Pathology and Imaging Findings
The lobar form of HPE is the most developed and least anomalous form. The cerebral hemispheres are rather well developed and separated (including thalamic nuclei) (38). The interhemispheric fissure and falx cerebri separate nearly the entire cerebrum with the exception of the most rostral and ventral frontal lobes. Careful scrutiny reveals some degree of hypoplasia of the frontal lobes. The hippocampal formations are nearly normal, and the temporal horns are well defined, appearing normal or nearly normal. The olfactory bulbs, tracts, and sulci may be normal, hypogenetic, or absent. Although there is an apparent interhemispheric fissure, cerebral cortex may be found crossing the midline along nearly the entire anteroposterior axis of the brain. The anterior falx is usually dysplastic. The corpus callosum shows mild dysplasia of the genu (Fig. 4.47). The frontal horns show variable degrees of development. They may be extremely rudimentary or may appear nearly normal (Fig. 4.47). The septum pellucidum is always absent. If a dorsal cyst is present, it is usually small.
Fetal Imaging
Nowadays, in most cases, the second trimester fetal MRI allows to confirm and correctly classify HPE suspected by prenatal ultrasonography (Fig. 4.48). The typical midline anomalies are easily detected, associated cortical abnormalities may be missed on early fetal MRI due to the small size of the developing brain, or the physiologic hypogyration of the brain during gestation may prevent accurate diagnosis of cortical abnormalities.
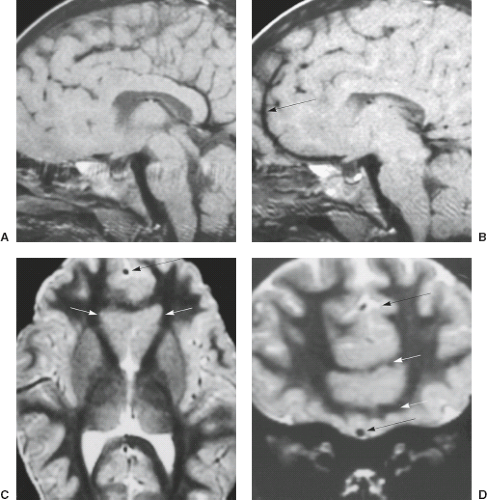 FIGURE 4.47 Lobar holoprosencephaly. A,B: Sagittal T1-weighted images show essentially normal callosal splenium and posterior body. Anterior body, genu, and rostrum, however, are not clearly visualized. This feature should prompt diagnosis of holoprosencephaly. Note the azygos anterior cerebral artery (long arrows). (C) Axial and (D) coronal T2-weighted images show abnormal gray matter in place of callosal genu (C, short arrows), complete coaptation of frontal horns, and two distinct white matter bridges of hemispheric fusion (D, short arrows). Note the azygos anterior cerebral artery (long arrows), which is far more anteriorly situated than in normal patients due to hypoplastic development of interhemispheric fissure. (From Truwit C, Lempert T. Pediatric neuroimaging: a casebook approach. Denver, CO: DPS Press, 1991, with permission.) |
 FIGURE 4.48 Semilobar holoprosencephaly as detected by fetal MRI at 23 weeks of gestation. (A) Sagittal and (B,C) coronal/axial T2-weighted images at 23 weeks of gestation show a single-lobed forebrain, fused central gray matter, absent septum pellucidum, partial development of the occipital and temporal horns, and a hypoplastic posterior falx cerebri. This imaging pattern suggests a semilobar holoprosencephaly. (Reprinted with permission from Dill P, Poretti A, Boltshauser E, et al. Fetal magnetic resonance imaging in midline malformations of the central nervous system and review of the literature. J Neuroradiol 2009;36:138–146.) |
Advanced Imaging
Only few articles have reported on the DTI and FT findings in HPE. In a patient with semilobar HPE, large white matter tracts were shown that were not apparent on conventional MRI. These large tracts were symmetric, projected on the expected course of the fronto-occipital fasciculus, and were connected across the midline in the subfrontal region. In addition, the precommissural fornix could be identified, but appeared markedly thickened and was located dorsally to the fused caudate nuclei. The appearance and location of the precommissural fornix by DTI and FT are supported by neuropathologic studies. Moreover, the superior and inferior fronto-occipital fasciculi could not be distinguished. In addition, in patients with severe forms of HPE (mostly alobar HPE), the corticospinal tract (CST) does not extend past the brain stem into the spinal cord and the medial lemniscal (ML) tracts are not separated. There is a strong correlation between qualitative evaluation of the dimensions of CST and middle cerebellar peduncles (MCP) with HPE severity and neurodevelopmental outcome.
Middle Interhemispheric Variant of HPE or Syntelencephaly
Definition
Syntelencephaly or the middle interhemispheric variant of HPE (MIVH) is a rare, milder form of HPE first described in 1993. Unlike semilobar HPE, the interhemispheric fissure and falx cerebri are variably formed both posteriorly and anteriorly, with midline cortical continuity in the posterior frontal and parietal regions (39).
Clinical Features
The clinical findings are similar to patients with a lobar HPE.
Pathogenesis and Genetics
Mutations in ZIC2, one of the HPE genes, have been found in patients with MIVH. Interestingly, ZIC2 mutations causing MIVH are milder compared to mutations causing other HPE phenotypes. This suggests a phenotype–genotype correlation.
Neuropathology and Imaging Findings
In MIVH, hemispheric fusion does not occur at the rostral forebrain, but across the adjacent posterior frontal and parietal regions (Figs. 4.49 and 4.50). Additional neuroanatomical characteristics of syntelencephaly include normally formed genu and splenium of the corpus callosum, but an abnormal callosal body; incomplete separation of the caudate nuclei and thalami, but normal complete separation of the hypothalamus and lentiform nuclei; nearly vertical orientation of the Sylvian fissures, which are abnormally connected across the midline over the vertex of the brain; subcortical gray matter heterotopia or cortical dysplasia in about 30% of the patients; and abnormality of the anterior cerebral vessels with an azygous ACA (39). Heterotopic gray matter may be seen “inside” the expected course of the corpus callosum (Fig. 4.49). Unlike classic HPE, the anterior third ventricle, basal forebrain, and olfactory system may be normal, as in cases of semilobar and some cases of lobar HPE (39). In the series reported by Simon et al., 19% of patients had cerebellar abnormalities (39). The face and orbits, as well as the pituitary and hypothalamus, often appear normal (39).
Related posts:
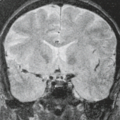

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


