Peptide
Receptor types/subtypes
Tumor expression
Somatostatin
sst1, sst 2 , sst3, sst4, sst5
Neuroendocrine tumors (gastroenteropancreatic tumors), lymphoma, paraganglioma, carcinoids, breast, brain, renal, small cell lung cancer, medullary thyroid cancer
Bombesin/GRP
BB1 (NMB-R), BB 2 (GRP-R),
BB3, BB4
Prostate, breast, pancreas, gastric, colorectal,
small cell lung cancer
VIP
VPAC 1 , VPAC2
Adenocarcinomas of breast, prostate, stomach and liver; neuroendocrine tumors
α-M2
α-M2-R
Breast cancer
α-MSH
MC1-5R
Melanomas
CCK/gastrin
CCK1, CCK 2
Medullary thyroid cancer, small cell lung cancer, gastrointestinal stromal cancer, stromal ovarian cancer, astrocytomas
Neurotensin
NTR 1 , NTR2, NTR3
Small cell lung cancer, colon, exocrine ductal pancreatic cancer, Ewing’s sarcoma, meningioma, astrocytoma, breast, prostate cancer
LHRH
LHRH-R
Prostate, breast, ovarian cancer
Substance P
NK 1 , NK2, NK3
Glial tumors (glioblastoma, medullary thyroid cancer), pancreas, breast, small cell lung cancer
Exendin
GLP-1
Insulinomas, gastrinomas, pheochromocytomas, paragangliomas and medullary thyroid carcinomas
RGD
α v β 3 -integrin
Glioma, breast, lung, prostate cancer
The biological actions of the peptides are mediated upon binding with high affinity to specific peptide receptors. Many of these receptors are highly overexpressed in a variety of human cancers, compared to their relatively low expression in normal tissues. This became the molecular basis for the development of radiolabeled peptides as radiopharmaceuticals for tumor imaging and peptide receptor radionuclide therapy (PRRT). After intravenous injection, the radiolabeled receptor-specific peptide will extravasate and bind to sites with high peptide receptor density, e.g., tumor. Diagnostic imaging and/or peptide receptor therapy follows depending on the radionuclide used for peptide labeling.
Peptide-based radiopharmaceuticals were introduced into the clinic more than two decades ago. The first and most successful registered diagnostic imaging agent to date is the somatostatin (SST) analog, 111In-DTPA0-octreotide (111In-OctreoScan) (Fig. 1). The U.S. Food and Drug Administration (FDA) approved 111In-DTPA0-octreotide which is effective for imaging SST receptor-positive tumors, such as neuroendocrine tumors (NETs), mammary cancer, and small cell lung cancer (Rufini et al. 2006; Ambrosini et al. 2011; Kaltsas et al. 2005). Another important application of radiolabeled octreotide and other SST-targeting peptide analogs is peptide receptor-mediated radionuclide therapy (Krenning et al. 1989; Kaltsas et al. 2005; Bodei et al. 2006; Teunissen et al. 2005). The thriving advent of 111In-DTPA0-octreotide raised interest in the development of radiolabeled peptides to target other tumor-related peptide receptors (Okarvi 2004; Fani et al. 2012a, b). This interest resulted in the discovery of bombesin analogs to target bombesin/gastrin-releasing peptide receptors (BN/GRPR), which are overexpressed in many common cancers including breast and prostate cancer (Reubi 2003; Gonzalez et al. 2008; Ohki-Hamazaki et al. 2005). Other radiolabeled peptides, such as vasoactive intestinal peptide (Hessenius et al. 2000; Raderer et al. 2000), neurotensin (de Visser et al. 2003; Buchegger et al. 2003), cholecystokinin/gastrin (Froberg et al. 2009; Kolenc-peitl et al. 2011), exendin (Wild et al. 2006, 2010a, b; Brom et al. 2010) and RGD (Arg-Gly-Asp) (Haubner et al. 2005; Decristoforo et al. 2008; Dumont et al. 2011), have been developed and are currently under preclinical or clinical evaluation to establish their applicability for the diagnosis or treatment of various cancers.
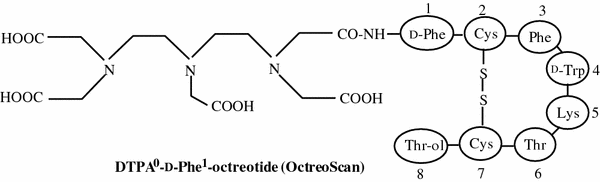
Fig. 1
Structure of DTPA0-D-Phe1-Octreotide
The term “theranostic” represents a combination of the terms “therapeutic” and “diagnostic”. Theranostics exemplifies the close association between diagnosis and therapy for personalized management of disease. The aim of theranostics in relation to nuclear medicine is to identify the appropriate molecular targets in neoplasms, so that the optimal ligands and radionuclides with favorable labeling chemistry can be selected that enables direct diagnosis and treatment specifically directed toward the individual tumor of the patient. Radiolabeled peptides targeting SST receptors, which are overexpressed in the majority of NETs, truly represent the essence of theranostics. Molecular imaging plays a key role in determining the success of radionuclide therapy. PET/CT modality using a 68Ga-labeled SST analog helps to select patients who are likely to benefit from peptide receptor therapy using β-emitters like 177Lu or 90Y labeled with the same SST analog. It also enables detection of primary and metastatic disease (staging), assessment of molecular response to therapy, and long-term follow-up after the initial diagnosis. In addition, dosimetry is achievable before or after therapy to ensure the optimum balance between risk and therapeutic benefit and to predict toxicity. The successful use of 68Ga for diagnosis, and of 177Lu and 90Y for radionuclide therapy using the same peptide for targeting SST receptor-positive NETs, has demonstrated the great potential of theranostics in modern nuclear oncology (Baum et al. 2012).
“Personalize” and “individualize” medicines have similar meanings. To personalize is to apply or understand as applied to a specific person. To individualize means to apply different strategies for different persons and to suit the requirements of an individual. Physicians have used the term “personalized” patient therapy for years. However, new technologies are now available (Denardo and Denardo 2012). Theranostics terminology has been used in the context of molecular targeting vectors (e.g., peptides) labeled either with diagnostic or with therapeutic radionuclides for the diagnosis and therapy of a particular disease, targeted specifically by the vector at its molecular level. Therefore, molecular imaging and diagnosis of the disease can be effectively followed by personalized treatment using the same molecular imaging vector. One of classic examples of theranostics in the personalized management of NETs, wherein the diagnosis, for example, using 68Ga-labeled SST analogs, is followed by personalized therapy using 177Lu-and/or 90Y-labeled SST analogs (Baum et al. 2012).
2 Characteristics and Limitations of Small Radiopeptides
The distinctive advantages of small radiopeptides over other biologically active molecules, such as proteins and antibodies, are summarized in Table 2. Receptors for peptides are often found in higher density on tumor cells than in normal tissues; hence specifically designed receptor-binding radiolabeled peptides could enable efficient visualization of tumors. Because of their small size, peptides usually exhibit rapid pharmacokinetics and good tumor targeting characteristics, with the ability to penetrate into tumors efficiently (Fani et al. 2012a, b; Langer and Beck-Sickinger 2001). As an advantage, the low molecular weight of the conjugate results in a fast diffusion rate. Peptides consist of a small number of amino acids (<50 amino acids) and can easily be prepared by standard solid-phase peptide synthesis (via convenient good manufacturing practices) and the desired pharmacokinetic characteristics can be molecularly engineered (by making appropriate changes in the peptide sequence) during synthesis and/or by adding a biomodifying molecule to the sequence (Antunes et al. 2007; Garcia et al. 2008). Automated peptide synthesizers are readily available for parallel synthesis procedures allowing the synthesis of peptide libraries in short time (Fani et al. 2012a). Peptides can also withstand harsher conditions for modification and labeling than antibodies, they are less likely to produce immunogenic response, and blood clearance and tumor uptake are faster. The faster clearance of non-target tissue leads to a better tumor-to-background ratio (better image contrast) and therefore to less irradiation of non-target organs (Fani et al. 2012b).
Table 2
Requirements of small peptides
Small in size |
Easy to synthesize |
Easy to radiolabel |
Feasibility of kit formulation |
Amenable to chemical/molecular modifications |
Ability to attach a chelating agent at the C- or N-terminus of the peptide |
Overexpression of peptide receptors in tumor cells and low expression in normal cells High receptor-binding affinity |
High tumor penetration |
Favorable pharmacokinetics |
Attain a high concentration in the target tissues |
Rapid clearance from the blood and non-target tissues |
Rate and route of excretion can be modified |
Few side effects |
Not immunogenic |
Many biologically important targets |
The binding affinity and the biodistribution are of major importance in the assessment of radiopharmaceuticals (Reubi et al. 1995). The first reflects the potential to fulfill its objective to target a receptor while the second is elementary for reaching the target and avoiding undesired radiation to non-target organs. Both are related to a major advantage of receptor-mediated peptide radiopharmaceuticals compared to, for example, conventional chemotherapeutics: the selectivity. Most receptors have high affinities for their ligands and are active at low nanomolar concentrations of the ligands. Therefore, radiopeptides with high specific radioactivity are crucial. Even small molar quantities of imaging agents may saturate a receptor, limiting the ability to visualize receptor expression and increasing the background due to non-specific binding. For biodistribution studies, tumor cells for which the bioconjugate is being developed are inoculated to animals (e.g., mice, rats) inducing the expression of a tumor. After administration of the radiopeptide, uptake in different organs, especially the receptor-positive organs and in the critical organs concerning toxicity effects may be determined.
For small size biomolecules, such as peptides the radioisotope label may significantly affect binding to the target receptor and in vivo pharmacokinetics. In this situation, the choices of radionuclide, labeling position, or location of the radiolabel may be important. The attachment of a bulky bifunctional chelator (BFC) to a relatively small peptide may lower also the binding affinity and biological potency. To reduce this negative influence, spacers between the peptide and the chelator may be introduced (Okarvi 2004; Fani et al. 2012a, b).
The biologic role of most receptors important in cancer is derived from their role in the tissue of cancer origin. In general, tumor receptors are expressed in the parent cell lineage and have an established physiologic function. For example, estrogen receptor expression is vital to the function of normal mammary gland epithelial cells. Many tumor receptors also play an important role in promoting carcinogenesis or tumor growth, as is the case for steroid receptors in breast and prostate cancer. The dependency on the receptor signaling pathway for tumor growth makes the receptor a suitable target for therapy, because interruption of the receptor-initiated signaling will result in a cessation of tumor growth and often tumor cell death. Thus, knowledge of the levels of receptor expression, which may vary significantly in different types of tumors and even in different sites in the same tumor, is required to infer the possibility that receptor-directed targeting will be effective (Mankoff et al. 2008).
One major problem associated with unmodified linear peptides is their often short biological half-life due to rapid proteolysis in plasma. As native peptides are degraded rapidly by endogenous peptidases and proteases before reaching the intended target, thus most peptides have to be modified synthetically to minimize rapid enzymatic degradation (Fani et al. 2012a; Behr et al. 2001). Great effort has been focused on developing metabolically stable peptides suitable for clinical use by performing appropriate molecular modifications, such as the use of D-amino acids for L-amino acids, the use of pseudo-peptide bonds, the inclusion of amino alcohols and the insertion of unnatural amino acids or amino acid residues with modified side-chains without compromising the receptor-binding affinity and biological activity of the peptide (Fani et al. 2012a). It is the specific amino acid sequence of a peptide and usually the nature and type of particular amino acid side-chains that determine resistance to enzymatic degradation. For example, native SST, which has a plasma half-life of ~2–3 min, but its molecularly modified synthetic peptide derivative, octreotide has a half-life of 1.5–2 h, making it suitable for clinical application (Mankoff et al. 2008).
Another issue related to radiolabeled peptides is often their high uptake and retention by the kidneys, which is of a concern, particularly for radiotherapy because of the potential nephrotoxicity (Teunissen et al. 2005; Virgolini et al. 2001; Valkema et al. 2005; Imhof et al. 2011). Though procedures have been developed and applied successfully in the clinics to manage nephrotoxicity that includes the infusion of a mixture of amino acids, such as lysine and arginine (Rolleman et al. 2003). There is a need to reduce kidney uptake and/or enhance renal clearance of the radiopeptide. Recently, it has been shown that the use of the cytoprotective drug amifostine (Rolleman et al. 2007a, b), and low doses of the plasma expander succinylated gelatin and gelatin-based gelofusine can inhibit the renal uptake of radiolabeled octreotide analogs (Vegt et al. 2006, van Eerd et al. 2006).
3 Development of a Peptide-Based Radiopharmaceutical for Tumor Targeting
The main steps involved in developing a radiolabeled peptide for successful receptor targeting are as follows: (i) identification of the molecular target (receptor), with relevance to human disease (unmet medical need), and search for a lead peptide, which may be a natural or a synthetic peptide, (ii) receptor density, homogeneity, and incidence are important factors in predicting successful in vivo tumor receptor targeting, (iii) solid-phase peptide synthesis (SPPS) of a tumor targeting peptide or its derivatives. In general, the design of a peptide is based on the structural composition of the endogenous ligand (natural peptide ligand), which exhibits high affinity for the corresponding receptor. The natural peptide molecule is often structurally modified to produce a metabolically stabilized analog, which preserves most of the biological activity and receptor affinity of the original peptide, (iv) covalent attachment of a chelating agent or a prosthetic group to the peptide either directly or through a linker/spacer group. The attachment of the chelating moiety to the peptide sequence may be performed at the non-active end of the main chain, mostly the N-terminus of the molecule, (v) radiolabeling that allows high labeling efficiency and high specific radioactivity, (vi) in vitro characterization, such as the binding of a radiopeptide with tumor cells, determination of receptor affinity, internalization into the tumor cells, and dissociation from the tumor cells, (vii) in vivo evaluation to assess the biological behavior, biokinetics, and tumor targeting capacity of the radiopeptide in animal models. Many aspects should be considered for further development, such as the accumulation of radiopeptide in target and non-target tissues, the rate and extent of the clearance of radioactivity from the body, the excretory pathway, and in vivo stability of the radiopeptide. (viii) Finally, the radiolabeled peptides, which successfully passed all the preclinical tests, after toxicological studies and established radiopharmaceutical preparation, may enter clinical studies in humans (Ambrosini et al. 2011). Tumor receptor targeting poses unique challenges for the design and development of a peptide-based agent. From the design of a new peptide molecule until the use in the clinical settings is a long way and from a large number of developed radiopeptides only few meet the criteria of a radiopharmaceutical for clinical application. The peptide-based radiopharmaceuticals in the field of targeted radiotherapy with impact in patient care are the SST receptor targeting analogs.
It is worth mentioning here that common to all receptors is the interaction of a ligand and the receptor, in which specific binding of the ligand to the receptor results in downstream biochemical or physiologic changes. Ligands that cause physiological functional changes upon receptor binding, typically the naturally occurring ligands, are called “agonists”. Ligands that bind to the receptor and block the binding of agonists but that do not activate changes are known as “antagonists” (Mankoff et al. 2008). It is generally believed that receptor agonist radioligands are more suited for tumor targeting as they exhibit good receptor-mediated internalization into the tumor cell upon binding to the respective cell-surface receptors, thus promoting active accumulation in the target results in optimal visualization (Langer and Beck-Sickinger 2001). Interestingly, however, a recent study performed with two potent SST receptor-selective antagonists demonstrated that the high affinity SST receptor antagonists that poorly internalized into tumor cells can also be effective in terms of in vivo tumor uptake characteristics in animal models as compared to the corresponding agonists, which highly internalized into tumor cells. This observation which was made both for sst2 and sst3-selective SST analogs, demonstrates that the SST receptor antagonists are preferable to SST agonists for in vivo tumor targeting (Ginj et al. 2006). In another study, 64Cu-CB-TE2A-sst2-ANT, a SST antagonist was evaluated for in vivo PET imaging of sst2-positive tumors and compared to 64Cu-CB-TE2A-TATE. The radioantagonists showed somewhat better pharmacokinetics over receptor agonists (Wadas et al. 2008).
The most recent study on the newly developed sst2-antagonist, namely LM3, demonstrated high and persistent tumor uptake of radiolabeled SST antagonists. Also, profound influence of the chelator and the radiometal was observed on the receptor-binding affinity of the radioconjugates (Fani et al. 2011). The first clinical study of SST antagonists confirmed the preclinical data as it showed higher tumor uptake of the antagonist 111In-DOTA-sst2-ANT compared to the agonist 111In-DTPA0-octreotide and improved tumor-to-background contrast, in particular tumor-to-kidney ratio (Wild et al. 2011a, b).
4 Development of Therapeutic Peptide-Based Radiopharmaceuticals
The introduction of 111In-DTPA0-octreotide (Fig. 1) for the diagnosis of SST receptor-positive tumors has opened the way to search for new receptor-based target-specific diagnostic and therapeutic peptides. The continued efforts have led to the research and development of several new peptide-based radiopharmaceuticals for the diagnosis and therapy of various human cancers as well as the discovery of several new chelating agents for peptide labeling with different therapeutic radionuclides (Fani et al. 2012a, b; Reubi et al. 2005).
Therapeutic radiopharmaceuticals are radiolabeled compounds specifically designed to deliver high radiation doses to receptor-specific disease sites (i.e., tumor lesions). The systemic noninvasive administration of radiopharmaceuticals that are designed for specific localization at tumor sites hold the great potential to treat disseminated disease. Most systemically administered therapeutic radiopharmaceuticals are small organic or inorganic compounds with definite structural composition. They can also be macromolecules, such as monoclonal antibodies or antibody fragments labeled with a therapeutic radionuclide. Ideally, the therapeutic radiopharmaceutical should localize at the tumor sites (even when their actual locations are unknown) in sufficient concentration to deliver a cytotoxic radiation dose to the tumor cells and clear efficiently from the blood and other normal tissues in order to minimize radiation damage to healthy tissues. In general, it was observed that radionuclide therapies have lower toxicities and longer term antitumor effects than conventional chemotherapy or external beam radiotherapy (Liu and Edwards 2001; Lee and Jeong 2012).
The utilization of radiometals in research and clinical practice has increased over the past 10 years and their radiochemistry has received renewed interest. One of the reasons is the development of medical radionuclide generators which produce positron emitting radiometals along with the tremendous advancement in medical imaging technology, such as the invention of PET/CT. But the main reason is the fast development of clinical targeted radiotherapy in nuclear oncology. In principle this technique can deliver high linear energy transfer (LET) radiation specifically to the tumor site. The search for an ideal therapeutic radionuclide for clinical use has been the driving force to produce new radionuclides with different half-lives and energy or even emitting different particles. Furthermore, efforts to produce some of the interesting lanthanides in carrier-free form appear to be successful and may lead to new therapeutic radionuclides. Of particular interest in this regard is the class of lanthanides including 90Y3+; several of them can be produced in high-flux reactors. As they have similar chemical properties, their production and use in therapeutic radiopharmaceuticals would allow studying the influence of dose rate and β− energy on tumor response as it can be assumed that the same radiolabeling chemistry can be applied to all lanthanide-based radiopharmaceuticals. This will most likely result in identical or at least very similar pharmacokinetic characteristics. (Fani et al. 2011b; Lee and Jeong 2012).
5 General Considerations for Therapeutic Radiopharmaceuticals
5.1 Choice of Therapeutic Radionuclides
Selection of the most appropriate radioisotope to be used for peptide receptor therapy is often a difficult task and need a careful consideration of several factors. These include: tumor uptake and retention, clearance from the blood and nontarget tissues, rate of radiation delivery, ease of radiolabeling, half-life of radionuclide, isotope availability, cost, and production method. An important characteristic of a therapeutic radiopharmaceutical is to deliver high radiation dose to the malignant cells with low damaging to normal cells (low side effects). The physical half-life of a therapeutic isotope should match the biological half-life of the radiopharmaceutical at the tumor site. If the half-life of radionuclide is too short, most of the dose will have been delivered before the radiopharmaceutical has reached the maximum target-to-background ratios. On the other hand, if the half-life of a radiopharmaceutical is too long it would cause unnecessary radiation burden to normal tissues. From a practical point of view, the half-life of the radionuclide should be long enough to allow sufficient time for manufacturing, release, and transportation to clinics (Liu and Edwards 2001). Some characteristics of therapeutic radionuclides commonly used for peptide labeling is summarized in Table 3.
Table 3
Characteristics of the commonly used radionuclides in peptide receptor therapy
Radionuclide | Type of emissions | Half-life | Maximum energy (keV) | Maximum tissue penetration range of particle | Production source |
|---|---|---|---|---|---|
Yttrium-90 (90Y) | β¯ | 2.67 days | 2284 | 12 mm | Generator |
Lutetitum-177 (177Lu) | β¯, γ | 6.68 days | 497 | 2 mm | Reactor |
Indium-111 (111In) | γ, Auger electrons | 2.8 days | 171 & 245 (γ) | 550 μm | Cyclotron |
3.6 & 19 (Au) | 10 μm | ||||
Copper-67 (67Cu) | β¯, γ | 2.6 days | 575 | 0.6 mm | Accelerator |
Bismuth-213 (213Bi) | α | 45.7 min | 5870 | 0.04−0.1 mm | Reactor |
For tumor receptor therapy, both α- and β-emitters are important. α-particles are particularly good cytotoxic agents because they disperse a high amount of energy within a few cell diameters. The short-range of α-emitters are more attractive if the radiopharmaceutical is internalized into cancer cells. A high LET and a short mean tissue penetration depth are the characteristics of α-emitters. But unlike β-emitters, α-emitters can effectively and irreversibly cleave DNA by causing direct double-strand breaks. So their therapeutic efficacies are less dependent on the effects of temperature, oxygen, and radiation sensitizers. In contrast, the cross-fire effects of α-emitters are expected to be lower than those of β-emitters due to their shorter mean tissue penetration depths. Although, the cytotoxicity induced by α-particles is far more selective and generally they have strong therapeutic effects and low toxicity, their clinical usefulness is limited due to several shortcomings. In particular, most α-emitters are not readily available and expensive. Also, their decay process involves several steps before forming stable isotopes. In addition, radiolabeled α-emitters tend to have lower chemical stabilities than β- or γ-emitters (Fani et al. 2011b; Lee and Jeong 2012).
In practice, an α-emitter intended for therapeutic use should have a moderate half-life, a suitable nuclear chemistry, and easy availability. One of the most important therapeutic α-emitters is 211At because of its relatively long half-life of 7.21 h. 211At is produced in a cyclotron using the 209Bi(α,2n)211At reaction, and can be collected by dry distillation method. It decays either to 211Po by electron capture or to 207Bi by α-emission. 211Po emits an α-particle to become 207Pb, with a half-life of 0.516 s, whereas 207Bi emits a β-particle to become 207Pb, with a half-life of 33.4 years. Thus, most of the therapeutic activity of 211At is exerted by α-particles. Another useful α-emitter is lead-212 (212Pb). Lead-212 has a half-life of 10.6 h. It decay chain includes the short-lived isotopes bismuth-212, polonium-212, and thallium-208, which all emit either α or β emission during decay over about another hour. At the end of the decay chain the stable element is 208Pb. Lead-212 decays to 212Bi by emitting a β-particle. 212Bi then decays to 208Pb by emitting two α-particles and two β-particles. Thus, 212Pb is often referred to as an in vivo generator because it produces more potent daughter radionuclides after administration. 212Bi has a short half-life of 60.6 min, but if it is administered as a 212Pb, its effect is extended to about 11 h. However, precautions are required concerning the purity and stability of the radionuclide-ligand complex after decay to 212Bi (McDevitt et al. 1998; Lee and Jeong 2012).
Recently, a radioimmunoconjugate containing the recombinant humanized monoclonal antibody trastuzumab conjugated with the bifunctional chelator TCMC ((1,4,7,10-tetra-(2-carbamoylmethyl)-1,4,7,10-tetraazacyclododecane), and radiolabeled with the α-emitting isotope 212Pb was evaluated as antitumor agent against HER2 expressing tumors. It has been shown that upon administration, 212Pb-TCMC-trastuzumab binds with high affinity to the extracellular domain of human epidermal growth factor receptor 2 (HER2); after internalization, the radioconjugate delivers a cytotoxic dose of α-radiation to the HER2-expressing tumor cells while sparing normal surrounding cells, due to the short path length of the α-particles. HER2, a tyrosine kinase receptor, is overexpressed on the cell surface of several cancers. The new agent, 212Pb-TCMC-trastuzumab has been authorized by the FDA to be administered to patients who have failed standard therapy and have HER2 expressing tumors mainly in the abdominal region.
Several potential isotopes are available but the radionuclides commonly used for labeling peptides include: yttrium-90 (90Y) and lutetium-177 (177Lu). 90Y has similar chemical properties and is usually being considered an analog of the lanthanides. 90Y (β¯, E max = 2.3 MeV, t 1/2 = 64 h) is the radionuclide of choice because of its high-energy pure β-particle emission with a 2.8 mm mean tissue penetrating range. 90Y is generator-produced and has a half-life of 2.7 days, which is short enough to achieve a critical dose rate but long enough to allow the radiopharmaceutical to be manufactured and delivered for clinical use (Fani et al. 2011a; Lee and Jeong 2012).
The in vivo dissociation and transchelation reactions of 90Y are the main sources of its toxicity. For example, reactions between 90Y chelates and biologically important metal ions, such as Ca2+ and Fe3+ produce free 90Y, which accumulates in bone and causes bone marrow toxicity. Competition between chelating moiety and native chelators present in the bloodstream, such as albumin and transferrin, may also result in early release of 90Y from 90Y-labeled bioactive conjugates. To circumvent these problems, DOTA derivatives, which form highly stable complexes, are better choices for 90Y than the less stable DTPA derivatives (Fani et al. 2011; Eisenwiener et al. 2000). To study the biodistribution and radiation dosimetry of 90Y, 111In(III) is often used as a surrogate, because 90Y does not emit γ-rays, which are required for diagnostic imaging in humans. The chelation chemistry of 111In with polyaminocarboxylates is similar to that of 90Y (Wilder et al. 1996).
177Lu is a relatively low-energy β-emitter and has a half-life of 6.75 days. 177Lu has a maximum energy of 0.5 MeV and a maximum tissue range of ~ 2 mm. It also emits γ-radiations of 113 keV (6.4 %) and 208 keV (11 %), which enables γ-scintigraphy thus facilitate dosimetry and disease staging. 177Lu is a reactor-produced radionuclide and is produced by irradiating enriched 176Lu. Similar to 90Y, 177Lu can also be complexed with DTPA and DOTA chelators. This is because 177Lu and lanthanide nuclides have the chemical properties that resemble those of 90Y. In particular, lanthanides and 177Lu produce more stable complexes with DOTA than DTPA.
Depending on the tumor size and location, the choice of the β-emitters may be different. As stated before, since high-energy β-radiation has a long penetration range in tissue, it is less efficient when treating smaller tumors (<1–2 g) as much of the energy is deposited outside the lesion. Therefore, high-energy β-particles (i.e., 90Y) are more appropriate for the treatment of larger tumors (with heterogeneous receptor distribution), whereas low-energy particles (i.e., 177Lu) may be more suitable for the treatment of small tumor lesions (Table 4). Indeed it has been shown that the combination of radionuclides with different β-energies and particle ranges may have better potential to achieve higher cure rates in tumors of variable sizes. Initial clinical studies have supported this assumption (Fani et al. 2012a; Kunikowska et al. 2011).
Table 4
Some of the major differences between 90Y and 177Lu-labeled radiopharmaceuticals
90Y-labeled radiopharmaceuticals | 177Lu-labeled radiopharmaceuticals |
|---|---|
Pure, high-energy β-emitter (2.27 MeV) | Medium-energy β-emitter (0.47 MeV), with low abundance γ-rays emission |
Half-life of 64 h | Half-life of 160.8 h |
Maximum tissue penetration range is 12 mm | Maximum tissue penetration range is 2 mm |
Shorter half-life allows to increased radiation dose | Smaller particle range and lower energy leads to better absorption in smaller tumors |
Higher energies and longer particle range, leads to more deposition of radioactivity in tumor cells | 177Lu emits γ-radiation suitable for γ-camera scintigraphic detection |
Better cross-fire through the tumor, which is useful in large/bulky tumor and heterogeneous distribution of peptide receptors on tumor cells | Longer half-life makes preparation of radiopharmaceutical and transportation to clinics more convenient |
Possibility of large absorbed radiation dose to the adjacent tissue, especially when the tumors are close to critical organs and normal tissues may receive large absorbed doses | |
Less suitable for smaller tumors because they will not be able to absorb all high-energy electrons emitteds |
5.2 Design of a Peptide Radiopharmaceutical for Tumor Receptor Targeting
Peptide-based targeted agents either design for diagnostic imaging or radionuclide therapy labeled with radiometals can be divided into four parts (Fig. 2):
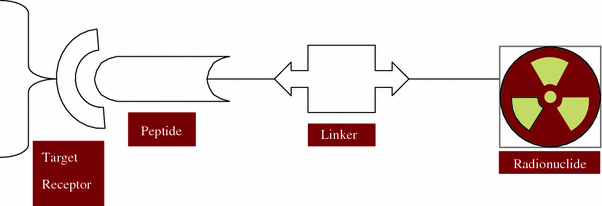
Fig. 2
Design of a radiolabeled peptide radiopharmaceutical for tumor receptor targeting
the peptide molecule which act as a target-specific vector,
the spacer/linker group,
the bifunctional chelator (BFC) and
the radiometal (i.e., 90Y, 67Cu, 111In, 177Lu, 212Pb, 212Bi, etc.).
The targeting molecules can be either large biomolecules like antibodies or small size peptide molecules. Between the targeting molecule and the radionuclide is the BFC, one end of which is covalently attached (conjugated) to the targeting molecule either directly or through a linker/spacer group while the other end strongly coordinates to the metallic radionuclide. A variety of BFCs are currently available for the strong attachment of metallic radionuclides and their selection mainly depends on the specific medical use (diagnosis or therapy). The choice of a linker depends on the pharmacokinetic requirements. They can be cationic, anionic, neutral, or metabolically cleavable molecules. In case of peptides, linker or spacer function is usually used to increase hydrophilicity in order to promote clearance through the renal route. While the nature of the vector molecule is determined by the cellular target (i.e., cell-surface receptor), the use of a spacer/linker group is often to improve the pharmacokinetics. The spacer group can also act as a separator between the BFC and the peptide to prevent steric influence of the chelator on the binding affinity of the receptor targeting part of the radiopeptide. The chemical nature of the radiometal determines the choice of the BFC (Fani et al. 2012a; Liu and Edwards 2001; Volkert et al. 1991).
Related posts:
 (Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
(Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
 Thyroid Carcinoma
Thyroid Carcinoma
 of Tumors: Pretargeting with Bispecific Antibodies
of Tumors: Pretargeting with Bispecific Antibodies
 Bisphosphonates for Bone Pain Palliation
Bisphosphonates for Bone Pain Palliation
 Radiopharmaceuticals for PET Dosimetry Before, During, and After Endoradiotherapy
Radiopharmaceuticals for PET Dosimetry Before, During, and After Endoradiotherapy
 Use of Dosimetry in the Planning of Patient Therapy
Use of Dosimetry in the Planning of Patient Therapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


