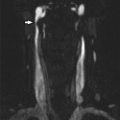
Fig. 1
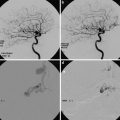
Conventional digital subtraction angiography of the left internal carotid arteries of a pediatric moyamoya disease patient. Anterior-posterior view (a–c) and lateral view (d–f) show steno-occlusive change of the terminal portion of the left ICA and moyamoya vessels in the perforator artery of the basal ganglia
Epidemiological Features
Geographical Distribution
East Asian countries feature the highest incidence of moyamoya disease, more particularly Japan and the Republic of Korea. A study conducted in Japan reported the total annual number of moyamoya disease patients at 3,900, with prevalence and annual incidence rates of 3.16 and 0.35 per 100,000, respectively, in 1994. The female-to-male ratio was 1.8 and 10 % of the patients had a family history of the disease. The distribution of the age at onset has two peaks: a dominant peak at 5 years of age and a more modest peak around the late 20s to 30 years of age [4]. A recent review of regional differences shows that incidences per 100,000 patient-years ranged in Japan from 0.35 to 0.94 (95 % CI 0.69–1.19) and in the USA from 0.05 (−0.04–0.12) in Iowa to 0.17 (−0.06–0.40) in Hawaii and were 0.41 (0.28–0.54) in Nanjing, China, and 0.02 (0.003–0.04) in Taiwan. The female-to-male ratio ranged from 1.1 (0.9–1.5) in Nanjing to 2.8 (1.2–6.1) in Iowa. Proportions with cerebral hemorrhage as the initial presentation were 56 % in China, 52 % in Taiwan, 29 % in Hawaii, 21 % in Japan, and 10 % in Iowa. Patients with childhood onset presented most often with ischemia (more than 75 %) in all regions [5].
Gender and Age Distribution
MMD has two peak ages of onset, initially at 5 years of age (pediatric or juvenile type) and subsequently at 30–50 years of age (adult type). Peak age in male is 10–14 years and 35–49 years, whereas peak in female is 20–24 years and 50–54 years [6]. In 2003, the total number of patients treated in Japan was estimated at 7,700. The female-to-male ratio was 1.8, and a family history was found in 12.1 % of patients. The prevalence rate was calculated at 6.03 per 100,000, and the annual rate of newly diagnosed cases was 0.54 per 100,000. In 2009, an analysis of the regional all-inclusive epidemiological data obtained in Hokkaido, a major island of Japan counting a population of 5.63 million [7], reported a prevalence of 10.5 patients per 100,000 and an annual incidence of 0.94 per 100,000, both of which greatly exceeded the results of the previous surveys. The female-to-male ratio was 2.18 and the age of onset occurred in two peaks with a different trend: the highest peak arose between 45 and 49 years, and the second occurred between 5 and 9 years. A familial history was observed in 15.4 % of patients. These epidemiological features differ significantly from the data obtained in previous studies. However, the higher detection and prevalence reported may not reflect an actual increase in the incidence of moyamoya disease, but may rather be due to the increased availability of noninvasive diagnostic tools, such as MRI/MR angiography.
Moyamoya disease patients’ age distribution shows characteristic bimodal pattern, pediatric and adult patients. The precise mechanism for this bimodal distribution has not been determined. In the pediatric patients, the rapidly progressing maturation of the brain may precipitate deficiency of a balance with cerebral blood flow maturation, and as a consequence, patients progress to develop ischemic attack. However, still many things are unknown about pediatric normal development of cerebral blood flow. This lack of balance between maturation of the brain and development of cerebral blood flow might not be able to explain the full mechanism of the development of ischemic attack in pediatric moyamoya disease patients. In adult moyamoya disease patients, hemorrhagic symptom can occur more commonly than in pediatric patients. In adult moyamoya patients, prevalence of ischemic and hemorrhagic symptom is almost the same.
Another unsolved issue of moyamoya disease is why the number of patients between these pediatric and adult moyamoya patients is small. About this issue, ischemic symptom which commonly occurs in pediatric period may become unlikely to occur because a balance between brain maturation and cerebral blood flow will be achieved. After getting over this dangerous pediatric period, moyamoya disease patient may become symptomatic due to a decrease of cerebral blood flow by arteriosclerosis in the adult period.
Familial Moyamoya Disease
An epidemiological feature of moyamoya disease is the high incidence of familial occurrence, which accounts for about 15 % of patients [8]. In patients with familial incidence, the ratio of women to men was reported to equal 5.0, as opposed to only 1.6 in sporadic cases; the mean (SD) age of onset was 11.8 (11.7) years in familial cases vs. 30.0 (20.9) years in sporadic cases. Out of eight parent-offspring pairs, all were paired with the mother. While parents presented the onset of symptoms at the age of 22–36 (mean 30.7 [7.5] years), their offspring presented symptoms much earlier, within 5–11 years of age (mean 7.2 [2.7] years). This phenomenon is called clinical anticipation. Clinical anticipation was reported in triplet repeat disease; however, triplet repeat has not been reported in moyamoya disease patients. This indicates a strong genetic association with a female predominance in cases of familial moyamoya disease [9].
Pathogenesis of Moyamoya Disease
Cytokines
Most studies about cytokines in moyamoya disease patients have been performed by the examination of cerebrospinal fluid (CSF) sampled from the subarachnoid space during surgical therapy. Angiogenetic cytokines such as basic fibroblast growth factor (b-FGF), hepatocyte growth factor, and transforming growth factor (TGF) showed increased concentrations in the CSF as well as in other surgical specimens such as the arterial wall specimens and the meningeal specimens. Among these cytokines, angiogenetic cytokine b-FGF has been thought to be related with several findings in moyamoya disease: steno-occlusive lesion in the circle of Willis, the development of moyamoya vessels and dilatation of the cortical small arterioles, and angiogenesis after surgical indirect synangiosis therapy. Reportedly, b-FGF induces the proliferation of vascular endothelial cells, which may precipitate the stenosis of the major arteries. On the contrary, b-FGF has also an angiogenetic and dilatation effect on the small arteries, which may explain the development of moyamoya vessels and the dilated pial arterioles on the cortex in moyamoya disease [10].
The role of cytokine abnormality in moyamoya disease has not yet been totally determined. Elevation of cytokines in the CSF may simply reflect the response of cytokines to hypo-oxygenation status in moyamoya disease. In this scheme, elevated concentrations of cytokines may simply be a consequence of hypo-oxygenation but not a direct pathogenesis of moyamoya disease. Validation study for the elevation mechanism of cytokine levels will be necessary [10].
Genetic Factors
In cases of familial moyamoya disease, the gene loci 3p24-p26 and 8q23 have been identified using genome-wide analysis, while loci 6q25 and 17q25 were determined using chromosomal search.
Cases of unilateral moyamoya disease progressing to bilateral moyamoya disease have been well known, as well as progression of major artery stenosis on the contralateral side to the initial disease. It has thus been suspected that major artery stenosis, unilateral moyamoya disease, and bilateral moyamoya disease were among a spectrum of relational phenomena, based on genetic susceptibility. In addition, familial moyamoya disease has been known to be an autosomal dominantly inherited disease with incomplete penetrance, and various stages of the disease are thus observed in familial moyamoya disease patients. From these points, a combination of internal genetic factors and external environmental factors has been believed to be relevant for disease occurrence or progression. These mechanisms of moyamoya disease have been reported to be relevant with three heterogeneities, disease heterogeneity, genetic heterogeneity, and locus heterogeneity [11].
Recently, a genome-wide association study identified ring finger protein (RNF)213 (http://omim.org/entry/613768) as the first moyamoya disease gene [12]. Also, another genome-wide linkage analysis by assuming the inheritance pattern of moyamoya disease as autosomal dominant mode with incomplete penetrance and whole genome-exome analysis provided evidence suggesting the involvement of RNF213 in genetic susceptibility to moyamoya disease [13]. Further studies are ongoing to clarify the biochemical function and pathological role of RNF213 in moyamoya disease [14].
Asymptomatic Moyamoya Disease
In recent years, asymptomatic cases of moyamoya disease and moyamoya disease manifesting with nonspecific symptoms only, such as headache, have been reported. “Asymptomatic” patients with moyamoya disease have been defined as those who have experienced neither ischemic nor hemorrhagic episodes since birth. According to previous reports, clues to the diagnosis of asymptomatic moyamoya disease included nonspecific symptoms such as tension-type headache, dizziness, head trauma, etc. Some patients were incidentally diagnosed using MRI/MRA which was performed for brain checkup or scrutiny for the disease because of family history of moyamoya disease [15]. The increase in the number of asymptomatic patients could be attributed, at least partially, to the current increasing availability of MRI/MRA examination in Japan. In an all-inclusive survey of moyamoya disease in Hokkaido, the authors reported that asymptomatic patients comprised 17.8 % of the 267 newly registered patients with moyamoya disease between 2002 and 2006 [7]. From this observation, the prevalence of asymptomatic moyamoya disease may be much higher than previously thought [16].
The reported imaging finding in asymptomatic moyamoya disease patients has been limited in number. Kuroda et al. reported cerebral infarction in 16 out of 77 (20.8 %) involved hemispheres in asymptomatic moyamoya disease patients [15]. About 40 % of the involved hemispheres had a moderate or severe reduction of cerebral perfusion reserve in asymptomatic moyamoya disease [15]. The annual risk for ischemic or hemorrhagic stroke was estimated at 3.2 %. Disease progression was associated with ischemic events or silent infarction in four of five patients. Also, asymptomatic abnormal findings were identified in another three patients [15].
Asymptomatic moyamoya disease may thus be closely related to the development of asymptomatic cerebral infarction. This observation may be consistent with the findings that steno-occlusive arterial change in adult moyamoya disease patients may progress in both the anterior and posterior circulation, in both bilateral and unilateral disease types, and in both symptomatic and asymptomatic patients [17]. The natural course of asymptomatic moyamoya disease is still not fully understood [3]. Therefore, it is important to know that moyamoya disease progression may occur asymptomatically and can suddenly cause ischemic or hemorrhagic stroke even in asymptomatic patients. It is currently unknown how and when moyamoya disease deteriorates from an asymptomatic to symptomatic. It might be feasible to monitor asymptomatic moyamoya disease patients by less invasive imaging methods such as MRI/MRA for disease progression and ischemic and/or hemorrhagic stroke findings [16, 18].
Recently, in Japan, asymptomatic moyamoya registry project (AMORE) has been started. By achievements of this study, progression mechanism and the natural course of asymptomatic moyamoya disease might be elucidated (Fig. 2).
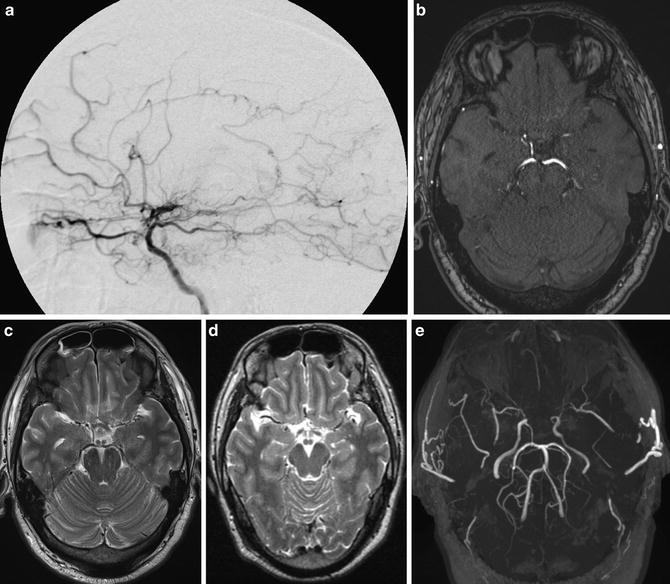
Fig. 2
A case of asymptomatic adult moyamoya patient. Conventional digital subtraction angiography lateral view image shows stenosis of ICA and moyamoya vessels at the frontal area and basal area. (a) Source image of TOF-MRA (b) and T2-WI image (c) shows both stenosis of ICA. Previous T2-WI image (d) shows apparent flow void of bilateral MCA compared to (c). MIP image of TOF-MRA shows bilateral ICA stenosis apparently (e)
Unilateral Moyamoya Disease
Based on the current diagnostic guideline [3], unilateral moyamoya disease in adult patient is diagnosed as probable moyamoya disease and refers to the presence of unilateral steno-occlusive change of the terminal portion of the internal carotid arteries accompanied by the formation of moyamoya vessels around that region. These unilateral changes may occur concurrently with other underlying diseases, such as hyperthyroidism, intracranial arteriovenous malformation, Down’s syndrome, Apert syndrome, von Recklinghausen’s disease (neurofibromatosis type 1), postirradiation therapy of the brain, systemic lupus erythematosus, and Sjögren’s syndrome; in these situations, the patient’s condition is classified as quasi-moyamoya disease and not as unilateral moyamoya disease [3]. In children, unilateral moyamoya disease associated with stenosis of the terminal portion of the internal carotid arteries should be considered as definitive moyamoya disease and not as probable moyamoya disease [19].
The frequency of unilateral moyamoya disease was reported as 10.6 % within 2,635 patients with moyamoya disease, including initially diagnosed and re-diagnosed patients [3]. A family history is occasionally present in patients with unilateral moyamoya disease. An analysis of 15 families having a family history of moyamoya disease in 3 or more generations revealed 5 patients with concurrent unilateral moyamoya disease in addition to 43 patients with definitive moyamoya disease and suggested the possibility that the disease was inherited by the same autosomal dominant inheritance pattern. From this observation, unilateral moyamoya disease with a family history can also be considered as a subtype of moyamoya disease. In addition, unilateral moyamoya disease without a positive family history should be distinguished from definitive bilateral moyamoya disease [20].
The symptoms of unilateral moyamoya disease are the same as those of definitive bilateral moyamoya disease. Ischemic symptoms, hemorrhagic symptoms, concurrent cerebral aneurysm, involuntary movement, and headache may be noted.
The reported frequency of progression from unilateral to bilateral moyamoya disease varies from 10 % to 39 % [21]. Each study cohort consists of a limited number of patients; therefore, progression rate from unilateral to bilateral disease may be influenced by the number of patients. Progression to bilateral moyamoya disease may not only affect pediatric patients but also adult patients. The statistically significant risk factors for progression to bilateral disease have been reported to be the presence of equivocal or mild stenotic changes in the terminal portion of the ICA, the middle cerebral artery (MCA), or the anterior cerebral artery (ACA) of the contralateral side [22] (Fig. 3).
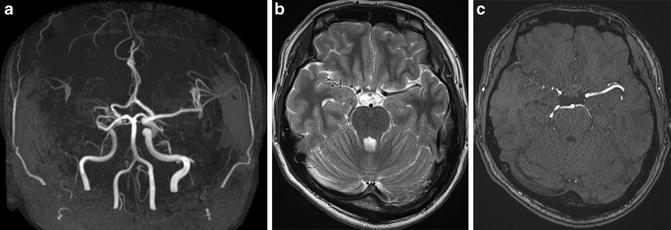
Fig. 3
A case of unilateral moyamoya disease patient shows right ICA stenosis on MRA. MIP image of TOF-MRA shows right ICA stenosis (a). Moyamoya vessels are not prominent in the cistern both on T2-WI image and TOF-MRA source image (b, c)
Quasi-moyamoya Disease
Quasi-moyamoya disease refers to the presence of steno-occlusive change of the terminal portion of the ICA accompanied by an abnormal vascular network in association with an underlying disease. Even in cases with unilateral lesions, if an underlying disease is present, the condition can be considered as quasi-moyamoya disease. Unilateral moyamoya disease in adult patient without underlying disease should be considered as probable moyamoya disease and should be differentiated from quasi-moyamoya disease. Quasi-moyamoya disease has been reported to affect people of all races. Concurrent occurrence with underlying congenital disease is more frequent in children; however, concurrent occurrence with acquired underlying disease is more frequent in adults [3].
The following illnesses have been reported as underlying diseases: atherosclerosis, autoimmune disease (systemic lupus erythematosus, antiphospholipid antibody syndrome, periarteritis nodosa, and Sjogren’s syndrome), meningitis, von Recklinghausen’s disease (neurofibromatosis type I), brain tumors, Down’s syndrome, head injury, irradiation, hyperthyroidism, stenocephaly, Turner’s syndrome, Alagille syndrome, Williams syndrome, Noonan’s syndrome, Marfan syndrome, tuberous sclerosis, Hirschsprung’s disease, glycogen storage disease type I, Prader-Willi syndrome, Wilms tumor, primary oxalosis, sickle cell disease, Fanconi’s anemia, spherocytosis, eosinophilic granuloma, type II plasminogen deficiency, leptospirosis, pyruvate kinase deficiency, protein S deficiency, protein C deficiency, fibromuscular hyperplasia, osteogenesis imperfecta, polycystic kidney, oral contraceptives, and poisoning (cocaine poisoning) [3].
Epidemiology of quasi-moyamoya disease was reported in the annual report of the Research Committee on Spontaneous Occlusion of the Circle of Willis (Moyamoya Disease) of the Ministry of Health, Labour and Welfare, Japan 2011 (http://mhlw-grants.niph.go.jp/niph/search/NIDD02.do?resrchNum=201128175A). Prevalence of quasi-moyamoya disease is reported as 5.4 % in a total of 7,941 moyamoya disease patients and 0.34 patients per 100,000 populations. Annual incidence of quasi-moyamoya disease is 0.11 patients per 100,000 populations.
Symptom of quasi-moyamoya disease may be epilepsy, headache, or asymptomatic. Concurrent presence of symptoms associated with mental retardation due to underlying disease and symptoms associated with cerebrovascular disorder may cause a relatively complicated clinical condition for the precise diagnosis. Cerebral conventional digital subtraction angiography can show various findings from those very similar to definitive moyamoya disease to be rather different, such as atherosclerotic lesions. Pathological findings also vary according to the underlying disease. Treatment of quasi-moyamoya disease is like that of definitive moyamoya disease. The effect of revascularization on the prevention of rebleeding in patients with quasi-moyamoya disease has not yet been clarified. In quasi-moyamoya disease, unilateral disease may progress to bilateral disease. The nature of the underlying diseases influences the prognosis of patients with quasi-moyamoya disease (Fig. 4).
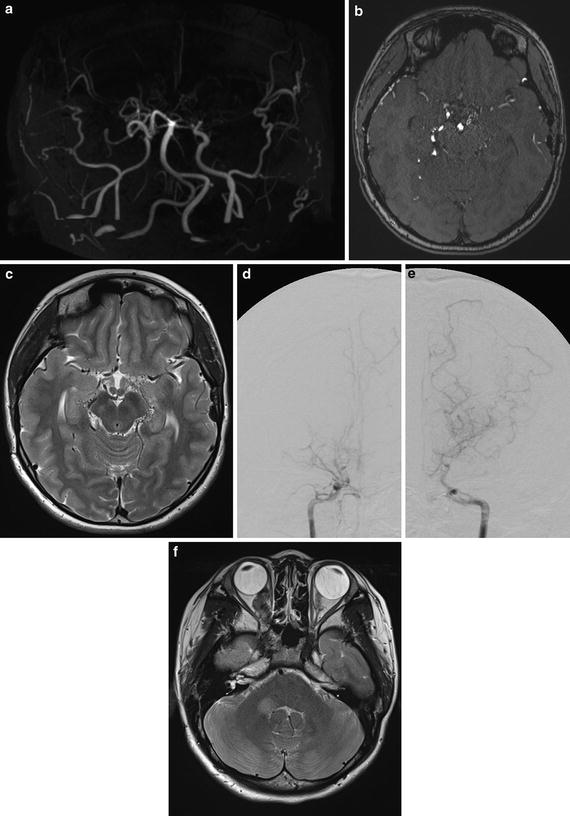
Fig. 4
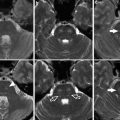
Images of a quasi-moyamoya disease (neurofibromatosis type I, von Recklinghausen disease) patient are shown. MIP image of TOF-MRA (a) shows steno-occlusive change of bilateral ICA. Source image of TOF-MRA shows cisternal moyamoya vessels as high-signal spots, which are also apparent on T2-WI image flow voids in the cistern. (b, c) Conventional digital subtraction angiography images (d, e) show steno-occlusive change and moyamoya vessels more apparently. On T2-WI image, right optic nerve mass lesion and high-signal area in the right cerebellar peduncle are apparent; those are characteristic lesion of neurofibromatosis type I (f)
Systemic Vessel Involvement of Moyamoya Disease
In addition to intracranial arterial steno-occlusive lesions associated with moyamoya disease, steno-occlusive lesions of extracranial arteries including renal, coronary, pulmonary, mesenteric, and peripheral arteries have also been reported [23]. Among these extracranial arteries, moyamoya disease most commonly affects the renal artery. The prevalence of renal artery involvement in moyamoya disease patients ranges from 5 % to 8 %. In most cases of moyamoya disease, in which the renal artery was involved, renal artery stenosis mainly involves the proximal one-third of the main branch. Severe stenosis case can precipitate renovascular hypertension. Although the definitive cause and pathogenesis of moyamoya disease remain unclear, pathologic studies of the steno-occlusive lesions of the cerebral vessels typically reveal fibrous thickening of the intima with a small amount of lipid deposition. Inflammatory cell infiltration is not noted in the vascular walls, and the internal elastic lamina is well preserved. Similar histological findings of intimal fibrous thickening in the extracranial vessels have been reported in autopsy cases of moyamoya disease [23].
Pathological Features
Pathological evaluation of involved vessels in moyamoya disease has shown that the outer diameters of the terminal portions of the relevant ICA are markedly diminished. In addition to outer diameter change, fibrocellular thickening of the intima, an irregular undulation (waving) of the internal elastic lamina, and attenuation of the media have been reported in a histological finding in the vascular wall of the ICA [24, 25]. Data from recent studies have suggested that caspase-3-dependent apoptosis might be associated with these histological changes in the vascular wall of moyamoya disease patients [26].
Moyamoya vessels consist of dilated perforating arteries, which can show a wide range of histological findings, including fibrin deposits in the wall, fragmented elastic lamina, attenuated media, and the formation of microaneurysms. In the early stage of the disease (stage I according to Suzuki’s angiographic classification), moyamoya vessels are rarely observed. Steno-occlusive change of the arterial lumen and subsequent thrombosis can also be seen in the moyamoya vessels [27]. Therefore, these histological changes might have close association with the onset or progression of ischemic or hemorrhagic stroke in moyamoya disease.
Clinical Features
An Overview
The clinical features of moyamoya disease show substantial difference between pediatric and adult patients. Most pediatric patients with moyamoya disease develop transient ischemic attack (TIA) or cerebral infarction, whereas about half of adult patients develop intracranial bleeding, and half develop TIA or cerebral infarction or both [28]. The symptoms and clinical course may change according to the age and the disease type manifested at the initial attack, and a wide range of severity of symptoms have been noted, such as TIAs and hemorrhagic stroke which resulted in permanent neurological deficits.
According to the recent increase in the availability of MRI/MRA, increasing numbers of patients have been reported, who were incidentally diagnosed as having moyamoya disease during the asymptomatic stage [15] or with only minor complaint such as headache [3]. In pediatric population, acute infantile hemiplegia is one of the most important symptoms of moyamoya disease. Benign familial nocturnal alternating hemiplegia of childhood has been also reported to be a possible symptom of moyamoya disease.
The frequency of each initial symptom in the 1,127 definitive moyamoya disease patients registered until 2000 is presented in Table 1 for the patients with the hemorrhagic type and ischemic type (infarction type, TIA type, and frequent TIA type) of initial attack. For both types, muscle weakness, consciousness disturbance, headache, speech disorder, and sensory disturbance were the most frequent; however, the incidence of consciousness disturbance and headache was higher. Incidence of muscle weakness was lower for patients with the hemorrhagic-type than for the ischemic-type initial attacks (p < 0.01) [3].
Table 1
Initial symptom of moyamoya disease (n = 1,127)
Initial symptom | Hemorrhagic type (%) | Ischemic type (%) |
|---|---|---|
Muscle weakness | 58.6 | 79.8a |
Consciousness disturbance | 70.4a | 14.1 |
Headache | 64.6a | 18.8 |
Seizure | 8.5 | 8.0 |
Psychiatric symptom | 8.7 | 2.5 |
Speech disorder | 24.5 | 20.1 |
Sensory disturbance | 18.4 | 19.3 |
Involuntary movement | 3.3 | 3.0 |
Intellectual disturbance | 5.3 | 6.2 |
Visual impairment | 2.0 | 3.2 |
Visual field defect | 3.9 | 5.0 |
Ischemic Symptoms
Moyamoya disease patients usually show cerebral ischemic change in the anterior territory of the ICA, particularly in the frontal lobe. Therefore, most patients with symptomatic moyamoya disease may present with focal neurological deficits, such as dysarthria, aphasia, or hemiparesis. However, moyamoya disease patients might also occur with other relatively atypical symptoms such as consciousness disturbance, sensory disturbance, visual symptoms, or involuntary movements, particularly in children [29, 30]. Some pediatric patients develop intellectual impairment owing to frontal lobe ischemia, infarction, or both [31]. In adult moyamoya disease patients, cognitive dysfunction, such as short-term memory disturbance, irritability, or agitation, has been reported. Patients who present with these atypical symptoms may be misdiagnosed with psychiatric disorders, such as schizophrenia, depression, or personality disorder.
In pediatric patients, ischemic attacks are typically induced by hyperventilation, for example, when crying or playing with a pinwheel. Electroencephalogram (EEG) findings in pediatric moyamoya patients are characteristic. In healthy children, high-amplitude slow waves are induced during hyperventilation, which will disappear after hyperventilation stops (an event known as build-up phenomenon). However, in pediatric moyamoya disease patients, the slow waves appear again after a recovery of normal ventilation. Pediatric moyamoya disease patients sometimes develop TIA when slow waves appear again during EEG scans. This reappearance of slow waves is pathognomonic for pediatric moyamoya disease patients and is known as rebuild-up phenomenon [32]. Recently, this rebuild-up phenomenon has been reported to be induced by post-hyperventilation hypoxia combined with a hyperventilation-induced reduction in cerebral blood flow and originates in the deep cortical sulci, where the cerebral perfusion reserve is diminished [33]. Rebuild-up phenomenon can completely disappear after effective surgical revascularization [33] (Fig. 5).
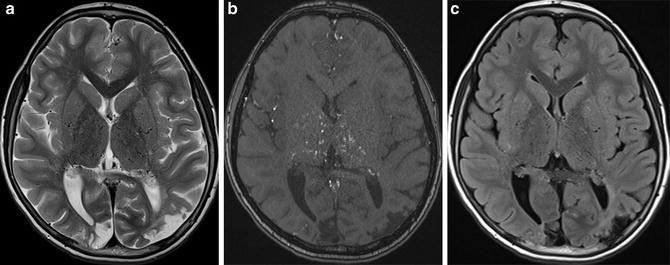
Fig. 5
3-T MR images of a pediatric moyamoya patient are shown. On T2-WI image (a), moyamoya vessels are apparent in the bilateral basal ganglia. On the source image of time-of-flight MRA (b), moyamoya vessels are apparent in the bilateral basal ganglia and thalamus. FLAIR image (c) shows tiny high-signal area in the bilateral occipital lobe (FLAIR ivy sign) and chronic infarction
Hemorrhage
About a half of adult moyamoya disease patients develop intracranial hemorrhage, though hemorrhagic symptom is rare in pediatric moyamoya patients [24]. Also, hemorrhagic symptom has been known to be more common in female patients than male patients. Hemorrhagic symptom patients show more severe symptom and deteriorated outcome than ischemic symptom patients.
Two main and one additional causes of intracranial hemorrhage in moyamoya disease have been assumed: (1) rupture of dilated, fragile moyamoya vessels or (2) rupture of saccular aneurysms in the circle of Willis especially in the posterior part. For the first cause, the rupture may be due to persistent hemodynamic stress on the moyamoya vessels and occurs mainly in the basal ganglia, thalamus, or periventricular region, which is common with basal moyamoya vessels. Intraventricular hemorrhage is more frequent in moyamoya disease patients than other origin hemorrhagic patients [34]. Peripheral aneurysms in the collateral vessels or moyamoya vessels might be identified on conventional digital subtraction cerebral angiography [35].
For the second cause, rupture of saccular aneurysms located around the circle of Willis occurs most commonly at the basilar artery bifurcation or at the junction of the basilar artery and the superior cerebellar artery. The vertebrobasilar system has an important role in providing collateral circulation for steno-occlusive change of bilateral internal carotid arteries in moyamoya disease patients. Consequently, hemodynamic stress probably may precipitate the formation of a saccular aneurysm predominantly in the posterior part of the circle of Willis or vertebrobasilar system. Rupture of a saccular aneurysm in the vertebrobasilar system can cause subarachnoid hemorrhage [36]. Therefore, in the imaging diagnosis of moyamoya disease patients, scrutiny for aneurysm especially in the vertebrobasilar system is important. 3-T MRI/MRA examination has superior signal-to-noise ratio, higher spatial resolution, and less invasiveness and, therefore, is suitable for clinical routine follow-up examination for moyamoya disease patients [18].
There is increasing reports that adult moyamoya disease patients might present subarachnoid hemorrhage over the cerebral cortex despite the absence of an intracranial aneurysm [37]. A third cause of intracranial hemorrhage in adult moyamoya disease patients is rupture of the dilated collateral pial superficial arteries on the brain cortical surface, although this is rare [37] (Figs. 6 and 7).
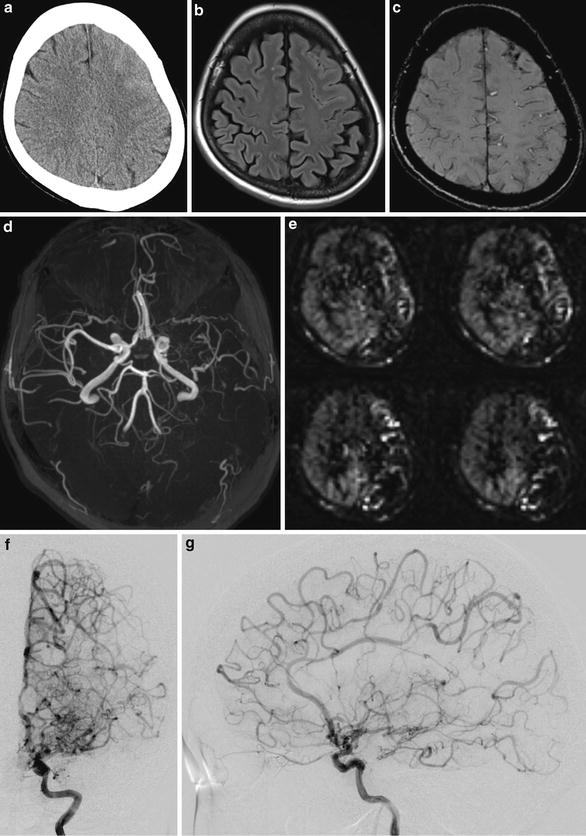
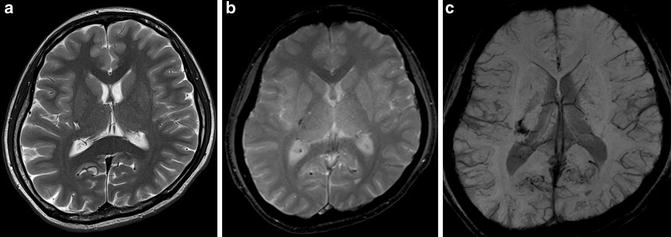
Fig. 6
In a case of patient presenting with headache, unenhanced CT scan image shows subtle high-density area in the left frontal lobe surface with obliteration of the left frontal lobe sulci. (a) FLAIR (b) and SWI (c) images show superficial high signal and low signal in the left frontal lobe, respectively. TOF-MRA MIP image shows stenosis of the left ICA and MCA (d). Arterial spin labeling perfusion-weighted image shows decreased flow in the left hemisphere. (e) Diagnosis of probable moyamoya disease was confirmed by a conventional digital subtraction angiography (f, g)
Fig. 7
Difference of appearance of hemorrhage among different MRI sequences. T2-WI image (a), T2-star-weighted image, and MiniP image of SWI of hemorrhage in the right basal ganglia are shown. T2-star-weighted image shows (b) hemorrhage as more pronounced (c) low-signal area compared to T2-WI image. This difference comes from effect of 180° refocus pulse used in T2-WI image. In addition, MiniP image of SWI shows hemorrhage as more pronounced low-signal area compared to T2-star-weighted image. This difference comes from the fact that SWI utilizes phase information to enhance the local susceptibility difference
Headache
Recently, headache is also considered to be one of the common clinical presentations of pediatric moyamoya disease patients. In many clinical cases, pediatric patients complaining headache may not be considered as having moyamoya disease, and other origins of headache such as meningitis or brain tumor will be looked for. Headache can be seen in about 20–30 % of moyamoya disease patients. Specific type of headache for moyamoya disease patients has not been reported; however, patients can show migraine in the more severely affected side. Also, patients with alternating hemiplegia of childhood showing migraine and hemiplegia have been reported [38]. Moyamoya disease patients may complain of headache before and after revascularization surgery. Preoperative headache in moyamoya disease patients was considered to be related to hypoperfusion or hypo-oxygenation because it has been reported that headaches disappeared after revascularization surgery [39].
Involuntary Movement
Involuntary movements are relatively rare symptoms in moyamoya disease patients, and their frequency is estimated about 5 % [40]. Association of various involuntary movement disorders with moyamoya disease has been reported in the literature. The spectrum of these involuntary movements includes chorea, choreoathetosis, dyskinesia, dystonia, limb shaking, and epilepsia partialis continua. Some of them may occur in a paroxysmal manner, while others can be triggered by the initiation of some movement (“kinesigenic” manner) [41], by some particular form of exercise, or by various types of hyperventilation maneuvers. The involuntary movements are usually transient, ranging in duration from several seconds to a few months, and rarely constant [40].
The main mechanism of onset of these involuntary movements is thought to be cerebral hypoperfusion induced by steno-occlusive change of the internal carotid arteries in the circle of Willis. Limb shaking is typically triggered by diffuse hemispheric hypoperfusion caused by severe stenosis of the ICA. Any activities inducing hyperventilation, such as singing and crying, may precipitate the involuntary movement, because of induced cerebral hypoperfusion. This hypothesis is supported by the fact that revascularization surgery is associated with improvement of the involuntary movements or at least a significant decrease in their frequency [40].
Diagnostic Evaluation
Current Diagnostic Criteria of Moyamoya Disease
1.
Cerebral angiography is considered essential for the diagnosis and must show at least the following findings [3, 42]:
i.
Stenosis or occlusion of the terminal portion of the intracranial ICA or proximal portions of the anterior and/or the MCA
ii.
Abnormal vascular networks in the vicinity of the occlusive or stenotic lesions in the arterial phase
iii.
Bilaterality of findings (i) and (ii)
2.
However, when magnetic resonance imaging (MRI) and magnetic resonance angiography (MRA) findings meet all of the following criteria, cerebral angiography can be omitted:
i.
MRA shows stenosis or occlusion of the terminal portion of the intracranial ICA or proximal portions of the anterior and/or the MCA.
ii.
MRA shows abnormal vascular networks in the basal ganglia.
Note: When two or more visible flow voids are present in the basal ganglia on MRI, at least unilaterally, they can be deemed as representing an abnormal vascular network.
iii.
Bilaterality of findings (i) and (ii).
3.
Moyamoya disease is an illness of unknown etiology. The differential diagnosis of this disease includes similar cerebrovascular lesions associated with the following underlying diseases, which should, therefore, be excluded: (i) atherosclerosis, (ii) autoimmune disease, (iii) meningitis, (iv) brain tumors, (v) Down’s syndrome, (vi) von Recklinghausen’s disease, (vii) head injury, (viii) cerebrovascular lesions after head irradiation, and (ix) others.
4.
Pathological findings that can be used as references for the diagnosis:
i.
Thickening of the arterial intima, mainly in the terminal portion of the internal carotid arteries, and narrowing or blockage of the lumen caused by this change, usually bilateral. Occasionally, lipid deposits are also present in the thickened intima.
ii.
Arteries such as the anterior, middle, and posterior cerebral arteries forming the circle of Willis occasionally show varying degrees of stenosis or occlusion associated with fibrocellular thickening of the intima, waviness of the internal elastic lamina, and thinning of the media.
iii.
Numerous small vascular channels (perforating and anastomotic branches) can be seen around the circle of Willis.
Reprinted with permission from Guidelines for Diagnosis and Treatment of Moyamoya Disease (Spontaneous Occlusion of the Circle of Willis). ©The Japan Neurosurgical Society [3].
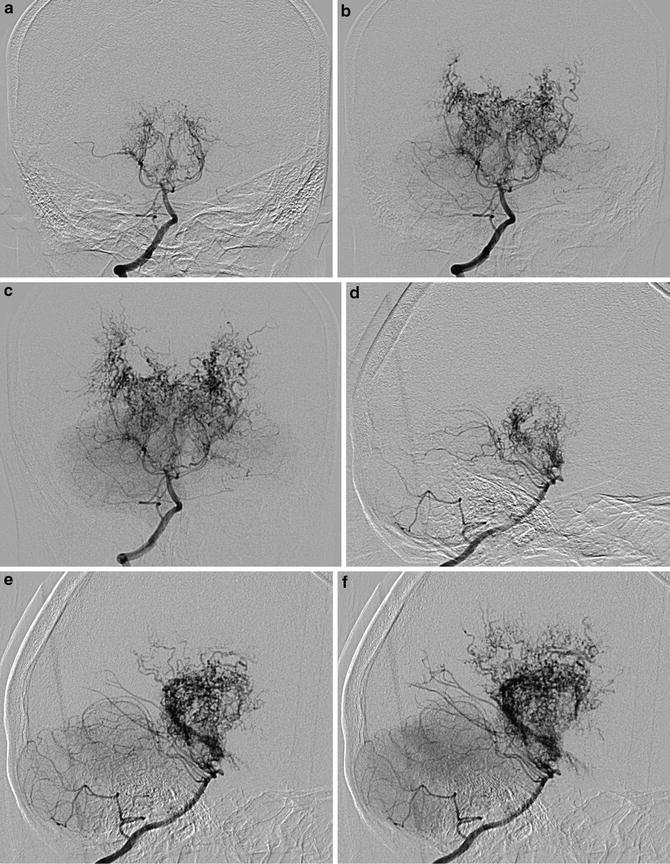
Fig. 8
In a pediatric moyamoya disease patient, right vertebral artery digital subtraction angiography images are shown. Anterior-posterior view (a–c) and lateral view (d–f) show collateral moyamoya vessels from vertebrobasilar systems
Definitive Moyamoya Disease and Probable Moyamoya Disease
Moyamoya disease should be classified as definitive or probable based on the abovementioned items (1) to (4) [3]. For definitive moyamoya disease, all criteria listed in (1) or (2) and in (3) should be fulfilled. In pediatric patients, however, the criteria in item (1) or (2) (i) and (ii), on one side, and visible stenosis around the terminal portion of the internal carotid arteries, on the other side, are sufficient for a definitive moyamoya disease diagnosis. For probable moyamoya disease, all criteria are fulfilled except item (1) (iii) and/or item (2) (iii) among the criteria of (1) or (2) and (3).
CT and CT Angiography in Moyamoya Disease
CT scan is one of the primary survey modalities for patients suspected of stroke; however, any definitive diagnostic information of moyamoya disease may not be obtained by this modality. In advanced stages of moyamoya disease, chronic cerebral infarction and unusual cerebral atrophy can be seen on CT scan; however, definitive diagnosis cannot be done [43]. On unenhanced CT scan, the steno-occlusive change and development of moyamoya vessels are not apparent. For the assessment of cerebral vessels, contrast-enhanced CT scan and CT angiography is necessary, and they can precisely show the steno-occlusive change and development of moyamoya vessels. During the diagnosis process of moyamoya disease, findings of unenhanced CT scan of stroke is not enough to exclude moyamoya disease, and further examination such as MRI/MRA or conventional angiography is necessary [43].
Unenhanced CT scan is a reliable imaging modality when the diagnosis of an ischemic or hemorrhagic stroke is suspected. In adult moyamoya disease patients, hemorrhage is common such as intraventricular hemorrhage, cerebral hemorrhage, and subarachnoid hemorrhage due to rupture of moyamoya vessels or aneurysms. In any cases of supratentorial cerebral hemorrhage seen on unenhanced CT scan, hemorrhage due to moyamoya disease must be considered as a differential diagnosis, and further examination such as contrast-enhanced CTA is recommended [43].
The recent advancement of multi-detector row computed tomography (MDCT) scanners has enabled high-resolution three-dimensional reconstruction. CTA and MRA were compared in terms of the steno-occlusive changes exhibited in each vessel. CTA and MRA scores were assigned on the basis of the severity of occlusive changes in the ICA, MCA, ACA, and posterior cerebral artery (PCA). CTA scores were significantly correlated with MRA scores, and their scores were in complete agreement in 39.6 %. The mean CTA score was significantly lower than the mean MRA score. CTA using MDCT is having a higher spatial resolution, therefore, can be a more reliable method than MRA for diagnosing moyamoya disease, particularly in emergency cases [44] (Figs. 9 and 10).
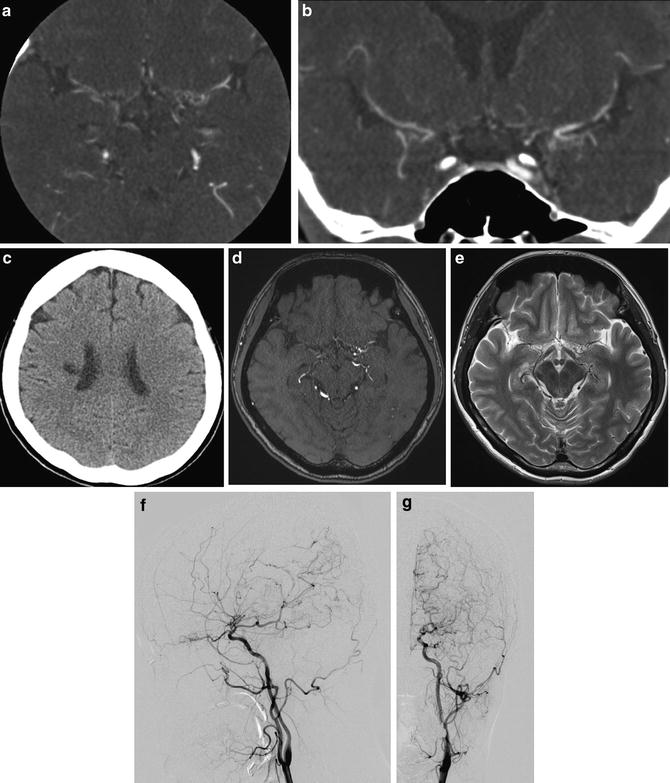
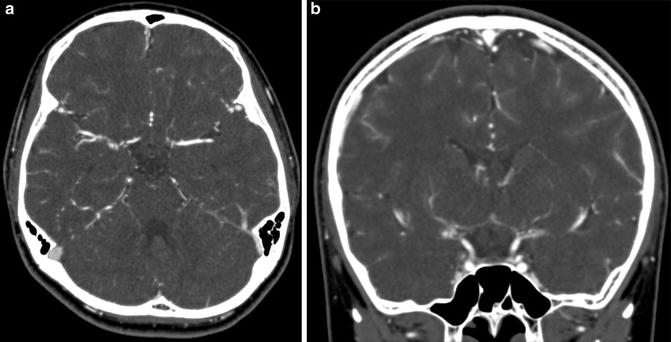
Fig. 9
CT scan image of an adult moyamoya patient shows chronic infarction as low-density area (c). Contrast-enhanced CT angiography images show moyamoya vessels in the cisternal area as tiny high-density spots. (a, b) In the same patient, MRA (d) and T2-WI images (e) show steno-occlusive change of bilateral ICA and moyamoya vessels. Final diagnosis was confirmed by conventional digital subtraction angiography (f, g)
Fig. 10
In a case of unilateral moyamoya disease, contrast-enhanced CT angiography image shows steno-occlusive change of the right ICA and surrounding moyamoya vessels in the cistern (a, b)
MRI/MRA Diagnosis of Moyamoya Disease
In moyamoya disease patients, anterior circulation (steno-occlusive change of the ICA and collateral moyamoya vessels), posterior circulation (steno-occlusive change of the PCA and leptomeningeal anastomosis collateral vessels from the posterior circulation), and transdural anastomosis collateral vessels such as ethmoidal moyamoya vessels and vault moyamoya vessels should be evaluated to monitor the hemodynamic state of moyamoya disease patients. Time-of-flight (TOF) MRA method is more suitable than phase contrast MRA or other MRA methods because TOF-MRA can show the steno-occlusive change of the major artery and development of moyamoya vessels simultaneously and more precisely. MRI/MRA examination can provide the significant diagnostic information less invasively (without ionizing radiation exposure) compared to conventional angiography.
Cerebral angiography is essential for a definitive diagnosis of moyamoya disease [3]. MRI/MRA can also be used for a definitive diagnosis when the following findings are fulfilled on TOF-MRA imaging conducted using a scanner with a static magnetic field strength of more than 1.5-T (especially 3-T MR scanner is preferable):
1.
On MRA, stenosis or occlusion of the terminal portion of the intracranial ICA or proximal portion of the anterior and/or middle cerebral arteries
2.
On MRA, abnormal vascular networks in the basal ganglia
Note: When two or more visible flow voids in the basal ganglia are present at least unilaterally on MRI, they can be deemed as representing an abnormal vascular network.
3.
Bilaterality of findings (1) and (2)
Moyamoya disease stage classification has also been reported based on the MR findings (Table 2). In this classification system, the stage is determined by simply assigning scores to the MRA findings and then totaling the scores. The stage classification using this method corresponds well to the conventional classification based on angiography and has been reported to show high sensitivity and specificity [45].
MRA findings | Score | |
|---|---|---|
Internal carotid artery | Normal | 0 |
Stenosis of C1 | 1 | |
Discontinuity of the C1 signal | 2 | |
Invisible | 3 | |
Middle cerebral artery | Normal | 0 |
Stenosis of M1 | 1 | |
Discontinuity of the M1 signal | 2 | |
Invisible | 3 | |
Anterior cerebral artery | Normal A2 and its distal | 0 |
A2 and its distal signal decrease | 1 | |
Invisible | 2 | |
Posterior cerebral artery | Normal P2 and its distal | 0 |
P2 and its distal signal decrease | 1 | |
Invisible | 2 |
MRA total score | MRA stage |
|---|---|
0–1 | 1 |
2–4 | 2 |
5–7 | 3 |
8–10 | 4 |
FLAIR Ivy Sign
Fluid-attenuated inversion recovery (FLAIR) image has been widely used for the diagnosis of stroke and other cerebral abnormalities with a high capability of lesion detection by nulling the CSF signal with an employment of sophisticated inversion pulse [46]. Characteristic imaging finding on FLAIR has been reported in moyamoya disease patients [46]. On FLAIR image, moyamoya disease patients may show linear or serpentine high signal in the sulci along the cortical surface, which is believed to represent dilated leptomeningeal collateral vessels [46], and this finding is called “FLAIR ivy sign” because of its appearance resembling an ivy on the wall. This sign is frequently observed in distal areas with decreased perfusion caused by vascular stenosis including moyamoya disease. This mechanism of high signal on FLAIR image may at least partly share with the mechanism of findings intra-arterial high signal on FLAIR ivy sign may partly share the mechanism of intra-arterial high signal observed in acute cerebral infarction on FLAIR [47]. The mechanism of FLAIR ivy sign is also applicable to the deep medullary vessel area in moyamoya disease patients, and linear high signal ivy sign was also reported in the deep white matter [48].
FLAIR ivy sign can change its appearance responding to the change of hemodynamic status and, therefore, is useful for the evaluation of hemodynamic status in moyamoya disease patients. Its presence correlates highly with reduced CVR indicating misery perfusion area, and regional arteriocapillary circulation time was elongated [49] but was decreased after bypass surgery, correlated with improved hemodynamic status observed by 123I-IMP-SPECT [50, 51] (Figs. 11, 12, 13 and 14).
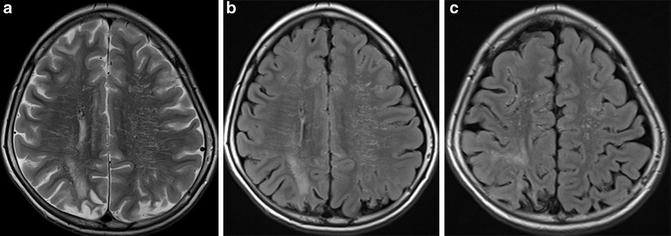
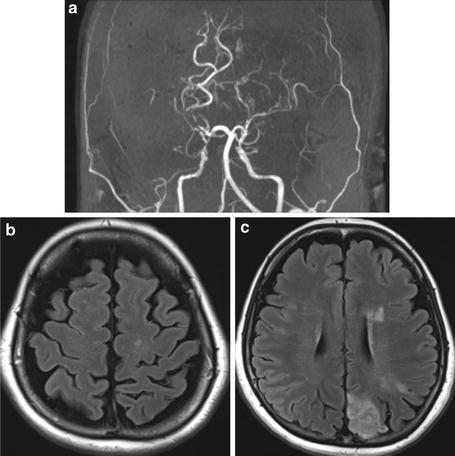
Fig. 11
3-T MR images of a pediatric moyamoya patient are shown. On FLAIR image (b, c), dilated collateral vessels in the cortical surface are noted as so-called FLAIR ivy sign. Also, collateral vessels in the deep white matter are noted (b). Infarction was noted in the bilateral parietal cortical area
Fig. 12




3-T MR images of an adult moyamoya disease patient are shown. (a) MIP image of TOF-MRA shows bilateral stenosis of ICA and moyamoya vessels. (b) FLAIR image shows intra-sulcal high-signal-intensity area representing dilated collateral vessels. This finding is known as “FLAIR ivy sign.” (c) Also, FLAIR image at lower level shows linear high-signal-intensity area in the deep white matter. This deep medullary vessel high signal on FLAIR also represents dilated collateral vessels. Infarction is apparent in the same slice
Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
