Fig. 22.1
Schematic presentation of: (a) graphene, (b) single-wall carbon nanotubes, (c) liposome, (d) core–shell nanoparticle, and (e) dendrimer
22.1.1.1 Graphene and Carbon Nanotubes
Graphene (Fig. 22.1a) is a 2-D sp2-bonded one-atom-thick carbon sheet in which carbon atoms are arranged in a regular hexagonal pattern. Graphene can be rolled into 1-D nanotubes (single-wall carbon nanotubes, SWNTs) which are just a few nanometers in diameter and several microns long (Fig. 22.1b). Graphene and SWNTs modificable chemistry and large surface area make them excellent candidates for the preparation of multifunctionalized nanosystems.
22.1.1.2 Vesicle-Type Systems
The development of vesicle-type systems like liposomes and micelles allows flexible mechanisms to deliver not only therapeutic-known drugs but other compounds of interest like contrast agents, antibodies and proteins that can be used for therapy and/or diagnosis. Liposomes (Fig. 22.1c) are small vesicles consisting of one or more concentric lipid bilayers enclosing discrete aqueous spaces. They are versatile in terms of size, surface charge, composition, bilayer fluidity, and are able to encapsulate drugs regardless of their solubility. Thus, they have been proposed for a long time as drug carriers with therapeutic purposes and also as potential imaging agents for the visualization of pathological processes.
Alternatively, recent biodegradable polymeric micelles have been developed as new vesicle systems that present remarkable drug delivery potential [13]. Polymeric micelles are formed by self-assembly of block polymers consisting of two or more polymer chains with different hydrophobicity. In aqueous environment, they form a core–shell micellar structure suitable for carrying poorly soluble drugs in its hydrophobic core. Moreover, its hydrophilic shell provides water solubility, protection, and functional groups suitable for further modifications.
22.1.1.3 Core–Shell Nanoparticles
Core–shell NPs (Fig. 22.1d) have a core surrounded by a hydrophilic shell and can be made of a wide variety of materials including quantum dots (QDs), metals, and metal oxides. Direct functionalization of core nanoparticles might be difficult, and, thus, they are often covered with a shell containing natural or FDA-approved biocompatible and degradable polymer(s) (see Table 22.1). An additional effect of embedding the core particle prevents degradation of the particle itself, thus, first, protecting its physicochemical characteristics and second alleviating potential safety concerns that might arise from trace ions. Due to size-dependent properties, low toxicity, large surface to volume ratio, and ease of fabrication and multifunctionalization, gold nanoparticles are the most commonly used in the biomedical field [14], although other materials like iron oxide have been also widely studied.
Table 22.1
Classification of biodegradable polymers
Biodegradable polymers | |||
|---|---|---|---|
Natural polymers | Synthetic polymers | ||
Protein-based polymers | Polysaccharides | Approved by FDA | Others |
Collagen | Agarose | Poly(glycolic) acid (PGA) | Poly(propylene fumarate) |
Albumin | Alginate | Poly(l-lactic acid) (PLA) | Tyrosine-derived polycarbonate |
Gelatin | Carrageenan | Poly(d,l-lactic acid) (PDLA) | Poly(glycolide-co-γ-caprolactone) |
Hyaluronic acid | Poly(d,l-lactic-co-glycolic acid) (85/15) (85/15 DLPLGA) | Ethylglycinate polyphosphazene | |
Dextran | Poly(d,l-lactic-co-glycolic acid) (75/25) (75/25 DLPLGA) | ||
Chitosan | Poly(d,l-lactic-co-glycolic acid) (65/35) (65/35 DLPLGA) | ||
Cyclodextrins | Poly(d,l-lactic-co-glycolic acid) (50/50) (50/50 DLPLGA) | ||
Poly(caprolactone) (PCL) | |||
Polyethylene glycol (PEG) | |||
22.1.1.4 Polymeric Dendrimers
Polymeric dendrimers (Fig. 22.1e) are hyperbranched nanostructures that can be controlled in size by controlling the number of polymerization generations. As polymerization progresses, a small, planar molecule transforms into a spherical nanostructure, with cavities where therapeutics and contrast agents can be grafted with great loading efficiency [15]. The majority of research into the use of dendrimers in the field of biomedicine has been concentrated on treatment, i.e., dendrimers delivering cytotoxic radiation via radioimmunotherapy. However, they have been eventually evaluated as diagnostic tools with potential applications in cancer.
22.2 Novel Imaging Probes
As outlined in the preceding chapters and section, molecular imaging techniques are in clinical use and applied in research. Hereby, differences between classical drug and contrast agent design on one hand and new, upcoming molecular structures have been described. The reader will be guided through individual aspects of the use of new molecular and imaging technologies in this section. For this, the reviewed examples for novel molecular imaging probes are mainly focused on preclinical in vivo applications in cancer diagnosis. If applicable, a reference to clinical applications has been made. Additionally, to cover the different imaging modalities, we deliberately choose to give each individual imaging modality a different weight. Nuclear imaging is already established, has its own established procedures to validate new molecular probes, and relies on radiochemistry expertise to radiolabel any structure. It has so done in the past, is doing at present and will continue to deliver new molecular probes in the future. New molecular probes in nuclear imaging are often prepared in situ and thus are described in detail, to reflect the special requirements of radiochemical synthesis. MRI was reduced to two applications: smart contrast agent and hyperpolarization of [1-13C]pyruvate. Hereby, smart contrast agents are of special interest as they have the potential to work as functional molecular imaging probes. Hyperpolarization for biomedical applications became recently available, and [1-13C]pyruvate has already reached the clinic as a potential imaging biomarker. Optical imaging, albeit an indispensable tool in cellular studies, was reduced to applications of QDs to cancer models, as they have the potential to build a versatile, sensitive biomedical imaging platform to cover microscopic, cellular, tissues, and in vivo applications.
22.2.1 Nuclear Imaging
Positron emission tomography and single photon emission computerized tomography are ultrasensitive, noninvasive molecular imaging techniques that allow the determination of the space-temporal distribution of a radiotracer after administration into a living organism. The main difference between them is that PET uses positron (β+)-emitting radionuclides whereas SPECT uses radionuclides that emit gamma (γ) rays. Annihilation of one positron with one electron produces two gamma rays, with equal energy (511 keV) and direction but emitted 180° from each other. Consequently, both techniques rely ultimately in the detection of gamma rays. Due to the high energy of gamma rays, they penetrate tissue almost unaffected and the detection is performed by using detectors located around the imaged organism. The measured photons allow the reconstruction of distribution of radioactivity as an image or sequence of images, which can be analyzed to get quantitative data (Fig. 22.2). Due to the high sensitivity, the radiotracer is usually administered in the sub-micromolar range, facilitating the study of biological systems without disturbing their function; this is, no toxicological, pharmacological, and/or undesired side effects are expected.
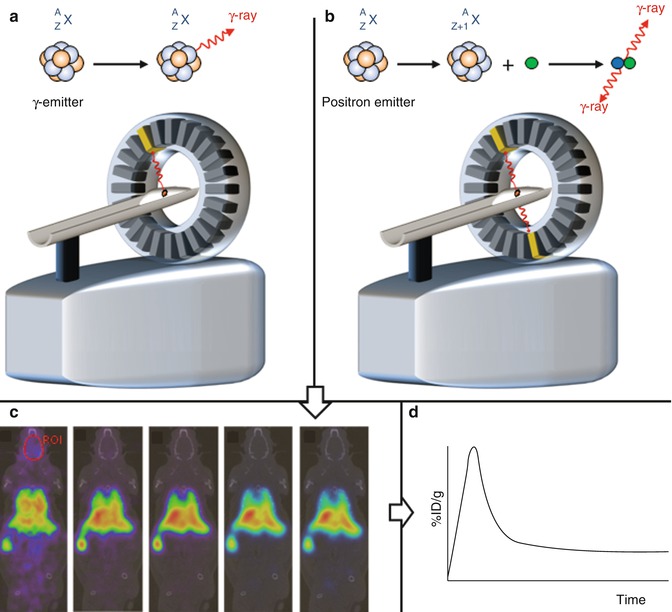
Fig. 22.2
Schematic representation of a single-photon emission computed tomography (SPECT) (a) camera and a positron emission tomography (PET) (b) camera. In SPECT, one disintegration produces one gamma ray. In the case of PET, the disintegration produces a positron, which annihilates with one electron producing two gamma rays, with equal energy (511 keV) and direction but emitted 180° from each other. In both cases, the detection of hundreds of thousands of photons allows the reconstruction of the distribution of radioactivity as an image or sequence of images (c). Regions of interest can be drawn and quantitative information can be obtained (d), e.g., as percentage of injected dose per gram of tissue (%ID/g)
The idea behind the application of nuclear imaging (PET and SPECT) for cancer diagnosis is therefore simple: A labeled species (radiotracer) which can provide information related to (1) a biological event (angiogenesis, apoptosis, hypoxia, proliferation, etc.) or (2) the presence of molecular biomarkers (protein kinases, growth factor receptors, specific overexpressed enzymes, or receptors, etc.) occurring uniquely or in a specific way in tumors must be synthesized and administered to the subject under study. Positron Emission Tomography or SPECT images will thus provide information related to the specific biological event or to the presence/absence of a particular biomarker. One classical example is 2-deoxy-2-[18F]fluoro-D-glucose ([18F]FDG), an analog of glucose with an 18F atom in the two position in substitution of an hydroxyl group. [18F]FDG is taken up by cells through the same pathways as glucose, phosphorylated and trapped in the cells (Fig. 22.3); as a result, [18F]FDG concentration increases in proportion to rate of utilization of glucose, and therefore, it can be used as an indirect proliferation marker; [18F]FDG is currently the most commonly used PET tracer in clinical diagnosis. A second example is [111In]pentetreotide (Fig. 22.4), the most widely used radiolabeled somatostatin analog, which binds to somatostatin receptors in the cell surface throughout the body. Tumors containing a high density of somatostatin receptors concentrate this radiotracer via receptor–ligand interaction.
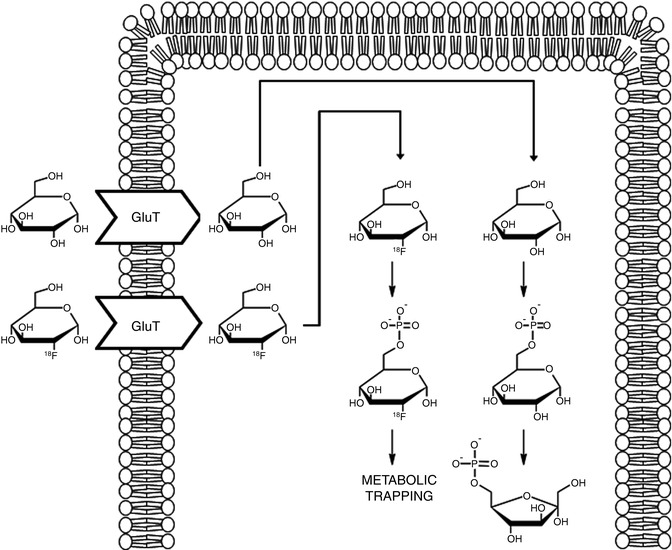
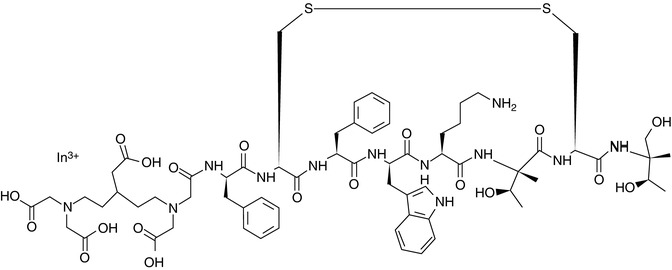
Fig. 22.3
Intracellular metabolism of glucose and [18F]FDG. Both are taken up by cells by means of glucose transporters (GluT). Once into the cell, they are phosphorylated to glucose-6-phosphate and [18F]glucose-6-phosphate, respectively. Unlike glucose, [18F]glucose-6-phosphate does not undergo further metabolism and is trapped in the cell
Fig. 22.4
Chemical structure of [111In]pentetreotide. 111In is complexed with the DTPA group on the left of the molecule
[18F]FDG and [111In]pentetreotide are only two examples of radiotracers which are currently used in the clinical practice. However, the vast amount of information resulting from biomedical research during the last years has enabled the identification of many molecular events that take place during disease processes. As a result, appropriate targets have been selected and a large number of labeled probes for the visualization of such processes in vivo have been developed. Indeed, hundreds of radiotracers based on a wide variety of radionuclides decaying due to β+ or γ emission have been developed and tested in animal models and/or clinical studies documenting their potential utility as molecular imaging probes. Among them, only a few have become radiotracers with diagnostic applications in the clinical environment. Table 22.2 collates some examples of radiotraces applied in oncology.
Table 22.2
PET and SPECT radiotracers: mechanism of uptake and localization
Radiotracer | Biochemical process | Mechanism of uptake or localization |
|---|---|---|
[18F]FDG | Glucose metabolism | Diffusion via glucose transporters. Substrate for hexokinase |
[18F]Fluoride | Bone metabolism | Incorporation of the hydroxyapatite crystals in bone |
[11C]Choline | Membrane synthesis | Substrates for choline kinase in choline metabolism |
[18F]Fluorocholine | ||
[18F]FMISO | Hypoxia | Intracellular reduction and binding |
[68Ga]DOTATOC | Receptor binding | Specific binding to somatostatin receptors |
[68Ga]DOTANOC | ||
[11C]l-methionine | Amino acid transport and protein synthesis | Transport into cells involves amino acid carrier protein. Intracellular trapping involves protein synthesis or transmethylation |
[64Cu]Annexin V | Apoptosis | Specific binding to phosphatidylserine on cell membrane |
[18F]FB-E[c(RGDyK)]2 | Angiogenesis | Integrin receptor on endothelial cells of neovasculature |
[99mTc]MDP | Bone metabolism | Incorporation of the hydroxyapatite crystals in bone |
[99mTc]HMDP |
A thorough description of all radiotracers used in clinical practice and their applications is far beyond the scope of this chapter. Instead, novel platforms or families of compounds that are currently explored in preclinical studies will be discussed and their labeling strategies utilized for their preparation will be presented. Two main goals lay behind the development of such new imaging agents: (1) the visualization of new molecular processes or biomarkers and (2) to gain specificity/selectivity in the visualization of such processes or biomarkers. Also, in an attempt to bring diagnosis and therapy closer, some newly developed imaging agents move towards the recently coined term theranostics. All these aspects will be covered and examples will be provided for clarity when appropriate.
22.2.1.1 Peptides
Generally, peptides considered as imaging agents have a low molecular weight, containing several to fewer than 50 amino acids. Small peptides have rapid clearance from blood and tissues, good permeability properties, and low toxicity. In addition, they can be usually chemically modified and incorporation of a radioisotope is feasible in most cases. Due to these facts, peptides have been extensively explored as diagnostic and therapeutic agents.
Most peptide-based PET and SPECT probes are designed on the basis of naturally occurring peptides. However, most of natural peptides have a short circulation half-life due to the action of peptidases and proteases. Therefore, identification of the active amino acid sequence and introduction of chemical modifications on the non-active amino acids sequence in order to improve in vivo stability (e.g., attachment of the peptide to a NP, substitution of one or more amino acids) is a commonly followed strategy. Just as an example, somatostatin (Fig. 22.5) is an endogenous peptide hormone that regulates the endocrine system and affects neurotransmission and cell proliferation. Certain subtypes of somatostatin receptors are overexpressed in certain tumor cells, and thus, ligands targeting such receptors may be good candidates to image a wide variety of tumors. However, somatostatin has a very short circulation time (2–3 min) [17], and thus, more stable analogs have been developed, like the abovementioned [111In]pentetreotide or 68Ga-labeled DOTATOC (Fig. 22.5). Additionally to improve stability, chemical modification in inactive sites is used to tune the excretion rate and permeability through membranes.
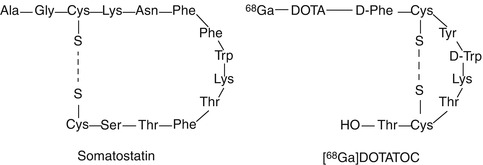
Fig. 22.5
Amino acid sequence of somatostatin and its analog [68Ga]DOTATOC
Radiolabeling of peptides can be achieved by following mainly three different strategies:
Radioiodination: Incorporation of a radioactive iodine atom (usually, 125I, 123I, or 131I and more recently 124I) is probably the most widely used alternative. Many options for such incorporation have been described in the literature; radioiodine can be incorporated by in situ oxidation of the anionic species (I–) using an oxidizing agent in solution, e.g., chloramine-T [18] and subsequent electrophilic substitution at the ortho– position to the hydroxyl group of a tyrosine residue. Eventually, the iodine atom can be introduced also in a histidine. This methodology provides high incorporation yields. However, not all peptides contain a tyrosine or histidine residue and, in some occasions, these amino acids (if present) might have a significant role in the biological activity of the peptide, which could be affected by the introduction of a bulky atom (notice that iodine is similar in size to a phenyl group and is markedly hydrophobic). Many investigations have also led to the conclusion that the loss of biological activity is dependent on the position of labeling and is sometimes unpredictable [19]. In addition, the peptide might not be stable under strong oxidizing conditions, e.g., oxidative reagents may cause oxidation of sensitive amino acids, like tryptophan to the oxindole and of methionine to the sulfoxide [20]. Finally, this labeling approach is very unspecific, because no control exists on the position in which the iodine atom is incorporated; the product mixture obtained is often complex and requires extensive high-performance liquid chromatography (HPLC) purification [21]. More convenient oxidizing agents have been developed to prevent oxidation of sensitive amino acids, like 1,3,4,6-tetrachloro-3α,6α-diphenyl glycoluril or Iodogen [22], a commercially available water-insoluble oxidizing agent which can be dissolved in an organic solvent and coated on the walls of the glass reaction tube. When Iodogen is used, the reaction can be terminated at any time by simply removing the crude from the reaction tube. Indirect methods, which consist of the covalent attachment of a pre-labeled group to one lysine, have been developed; the most widely used conjugation reagent is radioiodinated N-succinimidyl 3-(4-hydroxyphenyl)propionate (Bolton-Hunter reagent) [23]. However, in the indirect method multiple products can be formed by the reaction with the chain end and/or a variety of side chains, and this approach often requires an extensive purification of the resulting product. If the peptide under investigation already contains an iodine atom, isotopic exchange can be conducted [24]. A summary of the most commonly used strategies is schematized in Fig. 22.6.
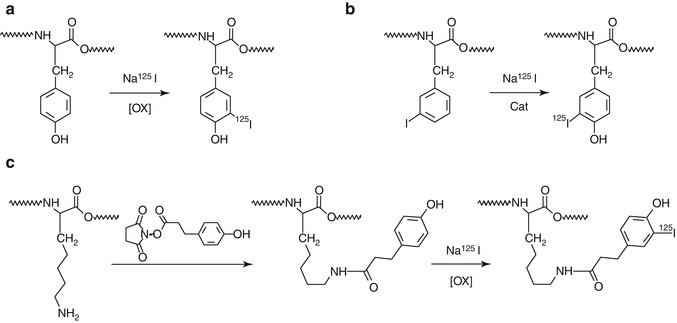
Fig. 22.6
Schematic representation of the main strategies used for the radioiodination of peptides; (a) electrophilic substitution, (b) isotopic substitution and (c) indirect labeling. Examples are shown with 125I; these strategies can be extended to other iodine radioisotopes
Radiofluorination: Incorporation of an 18F atom can be approached, in general terms, using two different strategies: (1) direct fluorination, in which 18F (as [18F]F– or [18F]F2) is introduced directly into the target molecule in a single step, and (2) indirect fluorination, in which a prosthetic group is used and multistep synthesis is usually required. Direct fluorination is rarely used in peptide chemistry. However, a large number of 18F-labeled prosthetic groups have been developed (Fig. 22.7), which are used for 18F-fluoroalkylation, 18F-fluoroacylation, or 18F-fluoroamidation of primary amino groups or thiol residues (see Fig. 22.8 for examples). More recently, 18F-labeled fluoroalkynes and azides have been developed and incorporated into proteins via the Huisgen cycloaddition of alkynes to azides (“click chemistry”) [25].
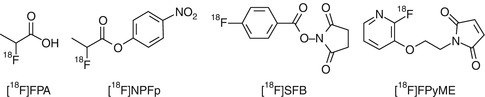
Fig. 22.7
Selected prosthetic groups for 18F labeling of peptides. FPA fluoropropionic acid, NPFP nitrophenyl fluoropropanoate, SFB succinimidyl fluorobenzoate, FPyME fluoropyridinyloxypropylmaleimide
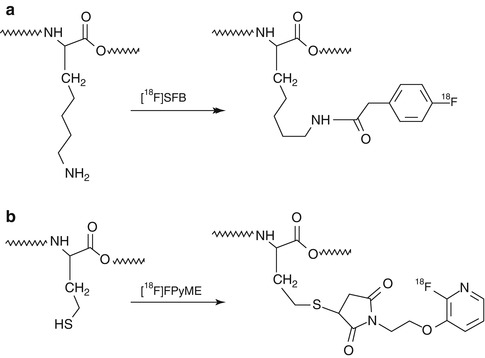
Fig. 22.8
Examples of incorporation of 18F into peptides using 18F-labeled prosthetic groups: (a) [18F]SFB and (b) [18F]FPyME
Incorporation of radiometals: This is usually achieved using bifunctional chelators (BFCs), which contain one functional group and a metal-binding moiety function. The first allows for anchoring the BFC to the peptide, while the latter enables the sequestration of the metallic radionuclide. This strategy has been successfully applied to the preparation of 64Cu-, 111In-, 68Ga-, 67Ga-, and 99mTc-labeled peptides, among others. Interestingly, this approach usually allows incorporation of the radiometal in the last synthetic step. In addition, most radiometals are commercially available or can be produced using generators. Hence, this strategy is becoming more and more popular, and the majority of recent publications related to peptide labeling involve the use of a radiometal.
As mentioned above, the vast amount of information resulting from biomedical research has enabled the identification of many molecular events that take place during disease processes, appropriate targets have been selected, and a large number of labeled probes for their visualization have been developed. Labeled peptides for the in vivo visualization of the most important targets associated to cancer include integrin receptors, somatostatin receptors (SSTR), gastrin-releasing peptide receptor (GRPR), melanocortin-1 receptor (MC-1R), vasoactive intestinal peptide receptors (VIPRs), glucagon-like peptide-1 receptor (GLP-1R), and neurotensin receptor (NTR). Extensive reviews covering the recent advances in specific delivery of peptide-based probes for PET and SPECT imaging have been recently published [26, 27], and therefore, only a couple of examples will be briefly presented here.
The first example involves the application of gastrin-releasing peptide (GRP) analogs to prostate cancer imaging [28]. In this work, [Lys3]bombesin ([Lys3]BBN) was conjugated with 1,4,7,10-tetraazacyclododecane1,4,7,10-tetraacetic acid (DOTA) and labeled with the positron-emitting isotope 64Cu. The labeled peptide was administered to PC-3 androgen-independent (AI) and CRW22 androgen-dependent (AD) prostate cancer tumor models. The peptide had a rapid blood clearance. At 1 h after administration, tumor-to-blood ratios were 13.1 ± 2.3 and 4.1 ± 1.3 for PC-3 and CWR22 tumors, respectively (see Fig. 22.9 for representative PET and autoradiographic images). These results indicated GRP receptor-specific uptake in PC-3 tumor, which was confirmed by in vivo receptor-blocking study at 1-h time point.
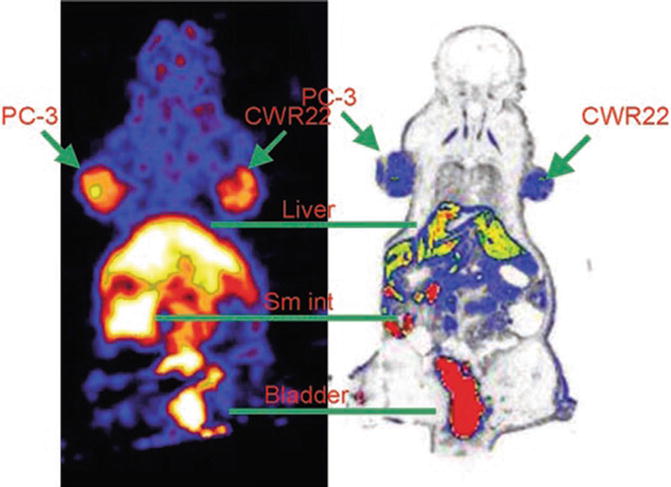
Fig. 22.9
(Left) Coronal microPET image of tumor-bearing mouse (PC-3 on left shoulder and CWR22 on right shoulder) 1 h after administration of 64Cu-DOTA-[Lys3]BBN. (Right) Digital autoradiograph of section containing tumors. Reprinted by permission of SNMMI from Chen et al. [28]
In another work, 4-([18F]fluoro)benzoyl-neurotensin(8–13) and two analogs stabilized in one and two positions were synthesized and evaluated in vitro and in vivo as potential candidates to image tumors overexpressing neurotensin receptor 1 (NTR1) by PET. In vitro binding affinity in the low nanomolar range to NTR1-expressing human tumors was found for all analogs. In vivo studies in rats and mice-bearing HT-29 cell tumors showed a moderate uptake of the radiolabeled peptides into the studied tumors, presumingly due to fast elimination by the kidneys. These results suggested that the high binding affinity to NTR1 and the stabilization against proteolytic degradation are not sufficient for tumor imaging by PET [29].
More promising results were obtained with an analog of glucagon-like peptide 1 (GLP-1), the natural ligand of GLP-1 receptors. The analog (exendin-3) was conjugated to DOTA for the incorporation of 68Ga and was investigated in vivo in BALB/c nude mice with subcutaneous insulinoma tumor cell (INS-1) tumors [30]. [Lys40(111In-DTPA)]exendin-3 was used as reference. A high uptake of [Lys40(111In-DTPA)]exendin-3 was observed in the tumor (33.5 ± 11.6%ID/g at 4 h after injection, see Fig. 22.10). Remarkably, tumor uptake of 68Ga-labeled [Lys40(DOTA)]exendin-3 was lower than tumor uptake of 111In-labeled [Lys40(DTPA)]exendin-3. This example highlights the potential effect of the BFCs and the radioisotope on the biological behavior of the radiotracers in vivo.
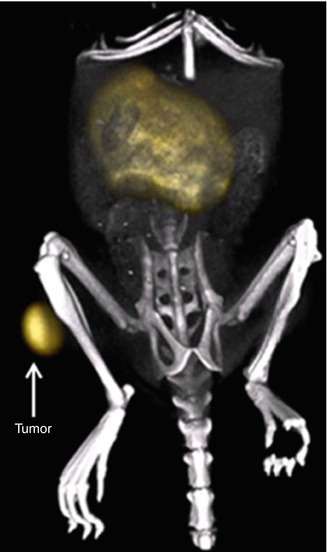
Fig. 22.10
Maximum intensity projection of a PET/CT image of a BALB/c nude mouse with a subcutaneous INS-1 tumor in the right hind limb after injection of 3 MBq HPLC-purified [Lys40(68Ga-DOTA)] exendin-3. The tumor (arrow) and kidneys are clearly visible. Due to the low background activity, the additional anatomical information given by the CT image helps to localize the tumor. Reprinted by permission of Springer Science and Business Media from Brom et al. [30])
22.2.1.2 Aptamers
At the beginning of the 1990s, it was observed that short ribonucleic acid (RNA) molecules could fold into three-dimensional structures and specifically bind different non-nucleic acid targets. Their surface was perfectly shaped to join them closely; hence, they were named “aptamers” (after the Latin aptus “joined”). Nucleic acid aptamers are capable of forming stable three-dimensional structures in aqueous solution and can be ribo- or deoxyribonucleic acids. In general, RNA offers the possibility of intracellular expression, whereas Deoxyribonucleic Acid (DNA) is more stable. No significant differences in specificity or binding abilities have been observed between these two types. Peptide aptamers, which are proteins that are designed to interfere with other protein interactions inside cells, will not be considered in this chapter.
Nucleic acid aptamers can be synthesized by Systematic Evolution of Ligands by Exponential Enrichment (SELEX) [31]. The SELEX procedure has been extensively described [32] and consists in a succession of 5 steps (See Fig. 22.11 for scheme) [33]. (1) An oligonucleotide library of sequences (candidates) is produced. All candidates contain two fixed sequences at their extremities to allow polymerase chain reaction (PCR) amplification, flanking a sequence of n nucleotides randomly synthesized. (2) The candidates are submitted to an in vitro selection based on their affinities for a specific target or a catalytic activity. (3) Sequences which satisfy the selection criterion are recovered while the others are removed. (4) The winning sequences are then amplified. Steps (2)–(4) are repeated several times. (5) Aptamers are cloned, sequenced, and tested for binding or catalytic activity.
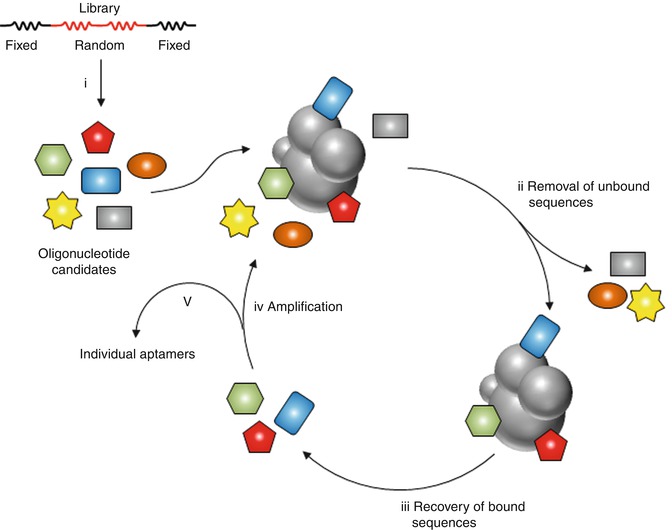
Fig. 22.11
General principle of SELEX. A random pool of oligonucleotides candidates is (i) synthesized and (ii) incubated with the target. Sequences satisfying the selection criterion are recovered while those that do not are removed. (iii) The bound sequences are extracted and (iv) amplified. The selected pool can then enter a new round of selection. (v) Aptamers are then cloned, sequenced and tested (Based on the drawing included in: Pestourie et al. [33])
Aptamers have been explored during the last decade as alternatives to monoclonal antibodies (mAbs). Monoclonal antibodies, because of their high affinity, specificity, and wide range of available targets, have been the workhorse of molecular targeting for long time. However, mAbs have a long blood residence, which decreases in vivo image quality. Interestingly, aptamers are comparable to antibodies in specificity and affinity for their target molecule [34]; they are intermediate in size (8–15 kDa) between antibodies (150 kDa) and small peptides (1–5 kDa) and are slightly smaller than single-chain variable-fragment antibodies (25 kDa). Aptamers readily support site-specific modifications that maintain structure and activity and can be coupled to diagnostic or therapeutic agents and to bioconjugates, such as polyethylene glycol (PEG) polymers, that can alter aptamer pharmacokinetics. They possess desirable storage properties, and elicit little or no immunogenicity in therapeutic applications. All these properties, in turn, make aptamers as good candidates to be labeled and used for the visualization of certain receptors in pathological processes. The most convenient way for the incorporation of a radionuclide into aptamers is based in the use of BFCs, which are covalently attached to a non-active site.
One of the first studies with aptamers for SPECT employed NX21909, an aptamer irreversible inhibitor of neutrophil elastase, which was successfully used for diagnostic imaging of inflammation in rats after being labeled with 99mTc [35]. First application to tumor localization came a few years later. TTA1, an aptamer to the extracellular matrix protein tenascin-C, was prepared and radiolabeled with 99mTc [36]. First, TTA1 was attached to the chelator 2-Mercaptoacetylglycylglycyl (MAG2) [37] through the 5′ amine for 99mTc incorporation. Scintigraphic images of U251 tumor-bearing mice were collected at different time points after administration. The images reflected high uptake in bladder and liver after 10 min; at 3 h, the intestines were prominent, with bladder still evident. After 18 h radioactivity had almost entirely cleared the body, and the tumor was the brightest structure visualized. Similar results were obtained with a breast tumor generated using the cell line MDA-MB-435 demonstrating the ability of the aptamer to recognize multiple tumor types. No uptake was observed in the tumor when a control aptamer was used (TTA1.NB). The accumulation in the tumors at different time points after administration can be clearly visualized in Fig. 22.12.
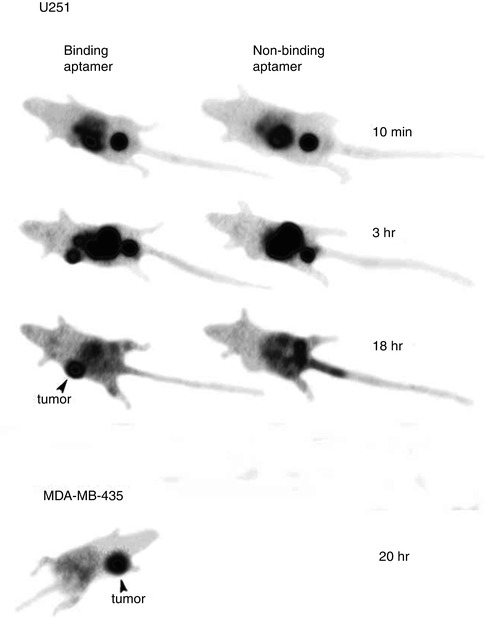
Fig. 22.12
Aptamer-based γ-camera images of tumors. With binding aptamer but not with control aptamer, U251 glioblastoma tumor is faintly visible at 10 min, prominent at 3 h, and the brightest structure at 18 h. Structures prominent at 10 min include bladder and visceral mass. Large intestine, bladder, and tumor are seen at 3 h. Also tested was MDA-MB-435 breast tumor implanted into mammary fat pad of female nude mice and allowed to grow to 400 mg. TTA1 was labeled with 99mTc, injected intravenously at 3.25 mg/kg, and imaged at 20 h. Reprinted by permission of SNMMI from Hicke et al. [36])
Results obtained with labeled aptamers have not been always successful. In a more recent work, selected aptamers against the protein core (AptA) or the tumor glycosylated (AptB) MUC1 glycoprotein were conjugated to MAG2 and labeled with 99mTc. The conjugation was achieved in high yield using standard coupling reactions between an amino modification on the aptamer and the activated carboxylic group on the ligands, and the biodistribution of both radiolabeled aptamers was studied in MCF7 xenograft-bearing nude mice at different time points after administration. The tumor uptake and clearance data showed a different behavior between the two aptamers, suggesting a quicker internalization in the tumor cells of AptB than AptA. However, the tumor/tissue ratios were not satisfactory, suggesting that these potential probes still need to be improved before being used as clinical radiolabeled targeting agents [38].
22.2.1.3 Antibodies
Antibodies are glycoproteins that can be made to specifically target the immunizing antigen. Thus, they have a huge potential as specific targeting agents for both diagnosis and therapy. Already in 1948 it was demonstrated that the immunoglobulin fraction containing antibodies to normal rat kidneys could be radiolabeled with 131I and used to specifically target the kidneys in vivo [39]. This work suggested the potential of antibodies for tumor imaging. Many years later, polyclonal antibodies to carcinoembryonic antigen showed that carcinomas can be imaged with radiolabeled anti-CEA antibodies [40]. Since these early works, labeled monoclonal antibodies have been used for imaging different types of tumors, including breast [41], colon [42], lung [43], ovarian [44], etc.
The utility of antibodies for imaging was initially limited by their large size (150 kDa). However, advances in antibody engineering have led to the development of various forms of antibodies, i.e., monovalent fragments (variable fragments, single-chain variable fragments), bivalent or bispecific diabodies, and minibodies. Additionally, improvements have been performed in modulating their immunogenicity, behavior in circulation, and pharmacokinetics. Finally, strategies for the efficient incorporation of radionuclides have been developed. As a result, antibodies and their modified forms have emerged as preferred agents to target tumor antigens. Currently, several antibodies have been approved by the FDA for imaging (reviewed in [45]). The recent advances in antibody-based oncologic imaging have been recently reviewed [46], and thus, they will not be further discussed in this chapter.
22.2.1.4 Nanocomposites
Due to the different chemical nature of the different NPs (see Sect. 22.1.1), many alternatives have been developed for the incorporation of the radionuclide. However, they can be summarized in 3 main categories:
Generation of the radioisotope by activation of the NPs, usually achieved by irradiation with ions (protons or deuterons) or neutrons. The radionuclide is thus generated in situ via a nuclear reaction. The activation of Al2O3 NPs by irradiation with 16 MeV protons via the 16O(p,α)13N nuclear reaction to generate 13N-labeled Al2O3 NPs [47], the activation of CeO2 NPs by deuteron irradiation via the 140Ce(d, p)141Ce nuclear reaction [48], and neutron activation of 197Au NPs to generate in situ198Au-labeled NPs [49] are only a few examples that can be found in the literature. Noteworthy, since neutron capture occurs preferentially at low neutron energies, it can generally be assumed that the activated nucleus remains very close to its original lattice position, this is, in the volume of the NP itself. This allows activation of NPs in solutions, whereas in the case of ion activation, radiolabeling occurs by recoil implantation of an activation product in a second NP, and thus, irradiation of nanoparticulate dry powders is required. In addition, by taking advantage of accelerator-driven neutron sources using the concept of Adiabatic Resonance Crossing, mild activation conditions (lower neutron and gamma-radiation dose rates and sample temperatures) allow the activation of nanoparticles or nanoparticle suspensions that are sensitive to thermal damage or radiation exposure [50–52].
After-loading or attachment/entrapment of the radionuclide once the NP has been synthesized. This is usually achieved using BFCs, which contain one functional group and a metal-binding moiety function. The first allows for anchoring the BFC to the NP surface, while the latter enables the sequestration of a metallic radionuclide. This strategy has been successfully applied to the preparation of 64Cu-, 68Ga-, and 99mTc- labeled NPs, among others [53–55]. One clear advantage of this methodology arises from the fact that NPs which already contain the BFC can be prepared and fully characterized. The chelation reaction usually takes place under very mild conditions, and thus, alteration of the NPs properties during labeling can be neglected. Alternatively, the attachment of pre-labeled tags to the NP surface can also be used [56]. Direct surface attachment of radioactive ions [57] and diffusion of radioactive species into the NPs have also been reported [58]. In the particular case of liposomes, transport of the radionuclide through the lipid bilayer and trapping in the aqueous interior are also possible [59].
Incorporation of the radioisotope during the NP synthesis process, using radioactive precursors [60].
In general terms, the second strategy is the most widely used in the biomedical field. The fact that the radionuclide can be incorporated in a very late stage in the synthetic process and the relatively mild reaction conditions required are definite advantages. Activation of NPs via neutron activation has been also occasionally used, although the number of available neutron sources is scarce, limiting thus its application. Activation of NPs using accelerated ions and incorporation of the radioisotope during the synthesis process have an interesting application field for the detection of inorganic NPs in environmental and toxicological studies.
Despite the number of scientific works reporting the labeling and subsequent utilization of nanoparticulate materials is continuously increasing, there are few concerns to be taken into account during the labeling process as well as during data interpretation. These concerns are not usually discussed in detail, but might lead to complete incorrect or misinterpreted results. Briefly (1) the term NP is never related to a single species, but to a group of species with very similar (but different) properties. In consequence, the macroscopic, measured biological readout is the sum of different individual actions, and, among those, only the ones derived from radiolabeled NPs will be detected and tracked. Thus, uniform or at least representative labeling must be guaranteed in order to get reliable data. (2) NPs are always altered to some extent during the labeling process. Hence, attention should be paid to characterization of the labeled NPs as well as to the assessment of their biological properties and compared to those of unlabeled ones; (3) nuclear imaging modalities actually track the radioactive atom, independently of its chemical form or environment. Thus, detachment of the radioisotope from the NP might lead to imprecise results and misinterpretation of the data; (4) the physical–chemical and structural properties of the unlabeled and labeled NPs are very similar, and separation of the second from the first is not usually feasible. Thus, the apparent specific activity is often low and the microdosing concept might be lost; (5) the labeling process and further in vivo detection of the labeled NPs require the manipulation of ionizing radioactive species and the use of sophisticated equipment which, in turn, makes the whole process complicated and expensive. These concerns have been recently discussed in detail [61].
Due to the abovementioned pitfalls, it is clear that despite radiolabeling results in an efficient way for the in vivo tracking of NPs after administration/incorporation to/into an organism, such issues need to be carefully considered if the results are to be valid. Noteworthy, the use of radiolabeled NPs becomes more difficult in the clinical environment. It is well known that radiotracers for human use need to be prepared under good manufacturing practices (GMPs), and during the last two decades, most of the radiopharmaceutical laboratories have updated their facilities and have implemented the procedures to accomplish the production under GMPs. However, production of labeled NPs under GMPs can be anticipated to be very challenging. All the equipment involved needs to be qualified. Importantly, because the radiotracer is usually released before sterility tests are finalized, the whole process requires full validation. In addition, radiotracers are usually autoclaved before release for sterility purposes or, alternatively, filtered through 0.22 μm filters. The stability of NPs to autoclaving conditions needs thus to be tested if filtration is not an option due to particle size. Finally, quality control can also become a big issue. Particle size distribution and chemical characterization are usually performed with equipment which is often outside radiation-controlled zones, and NP samples must be allowed to decay to acceptably low activity levels before full characterization can be achieved. All these questions and compliance with regulatory issues need to be carefully addressed before a labeled NP can enter a clinical trial.
Labeled NPs with Diagnostic Applications
As mentioned above, accumulation of NPs in tumor tissue may occur by the EPR effect; indeed, such effect has been exploited in certain occasions, and some examples can be found in the literature. Alternatively, tumor-specificity active targeting using NPs can be accomplished by including a target moiety attached to the surface of the NP, e.g., a ligand such as the RGD peptide [62], epidermal growth factor (EGF) [63], folate [64], transferrin (Tf) [65], or antibodies and antibody fragments, such as a single-chain variable fragment as discussed in previous sections; these moieties recognize a cell surface receptor which is only (or predominantly) expressed in tumor cells [66, 67]. In most cases, these ligand–receptor interactions result in efficient uptake of the NP into the tumor cell by receptor-mediated endocytosis. From this approach, an immediate question may arise: Why should one attach a targeting moiety to an NP, if the targeting moiety is capable to reach the tumor by itself, as explained in previous sections? Actually, there are different reasons that support the use of NPs as vehicles: (1) each NP can incorporate a high number of targeting moieties (multivalency effect); this, in principle, increases the probabilities of targeting moiety–target interactions and enables the simultaneous interaction of various targeting moieties with various binding sites, improving the effective accumulation of the NP in the targeted tissue; (2) NPs can be multifunctionalized; in other words, the targeting moiety and a therapeutic agent can be simultaneously loaded onto the NP, enhancing the selective accumulation of the therapeutic agent in the targeted tissue; and (3) by being attached to the NP, many bioactive molecules, which could be metabolized or rapidly eliminated, stay longer time in the circulatory system, resulting in an increased bioavailability. For example, attaching a peptide to an NP is expected to have a positive effect, with similar results to those achieved by chemical modification of the peptide. Worth mentioning, alteration of the biological activity due to attachment to the NP should be thoroughly investigated before application.
Given the variety of different cancer types and the multitude of nanoagents designed for their detection and/or treatment, it is impossible to enumerate all of them in this chapter. Thus, we will mention a few examples of some selected targeted and nontargeted nanoagents which have proven efficiency at least in animal models.
PEG-functionalized SWNTs containing c(RGDyK), a potent integrin αvβ3 antagonist, and a bifunctional chelator for the incorporation of 64Cu were studied as potential imaging agents in a mouse model bearing subcutaneous integrin αvβ3-positive U87MG tumors using PET [68]. The SWNTs were labeled with 64
 New Therapies and Functional-Molecular Imaging
New Therapies and Functional-Molecular Imaging
 Overview of Functional MR, CT, and US Imaging Techniques in Clinical Use
Overview of Functional MR, CT, and US Imaging Techniques in Clinical Use
 MRI Hypoxia Measurements
MRI Hypoxia Measurements
 Spectroscopy of Cancer
Spectroscopy of Cancer
 Current Clinical Imaging of Hypoxia with PET and Future Perspectives
Current Clinical Imaging of Hypoxia with PET and Future Perspectives
 Medical Image Computing for Oncology: Review and Clinical Examples
Medical Image Computing for Oncology: Review and Clinical Examples




Related posts:
New Therapies and Functional-Molecular Imaging
Overview of Functional MR, CT, and US Imaging Techniques in Clinical Use
MRI Hypoxia Measurements
Spectroscopy of Cancer
Current Clinical Imaging of Hypoxia with PET and Future Perspectives
Medical Image Computing for Oncology: Review and Clinical Examples
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
