Pediatric Brain Tumors
Robert A. Zimmerman
Larissa T. Bilaniuk
Introduction
Tumors of the central nervous system (CNS) in pediatric patients are the second most common form of neoplasia (1). They are exceeded only by hematologic malignancies such as leukemia. Brain tumors arising during childhood account for 15% to 20% of all primary brain tumors. The location of brain tumors in children has long been divided into those that are located infratentorially and those that are located supratentorially. Prior to the advent of modern neuroimaging with computed tomography (CT) and, more recently, magnetic resonance imaging (MRI), it was thought that infratentorial tumors were more common than supratentorial tumors (2). With the greater sensitivity of CT and MRI for the detection of small cerebral tumors, the incidence has become recognized as being equal, if not slightly in favor of the supratentorial tumors. Aside from location, the histology of pediatric brain tumors is quite different from that seen in the adult population. In adults, metastases, meningiomas, and malignant gliomas are the most frequent tumors. In the pediatric population, aggressive tumors of embryologic origin such as primitive neuroectodermal tumors (PNETs) are relatively frequent, as are low-grade glial tumors of astrocytic differentiation. Moreover, certain tumors that are not uncommon in the pediatric age group are exceedingly rare in the adult population. Hematogenously disseminated metastases are extremely unusual in the pediatric population, but dissemination throughout the subarachnoid space can be rather common, depending on histopathologic type.
The overall incidence of pediatric brain tumors is between 25 and 40 per million, with a male-to-female ratio of 1.2 to 1. Tumors of astrocytic origin comprise 40% to 55% of the total, or 16.8 per million; PNETs 20% to 30%, or approximately 5 per million; and ependymomas 10% to 15%, or approximately 2.2 per million.
Whereas most pediatric brain tumors appear to arise de novo without an underlying genetic cause or predisposition, certain genetic conditions do have a predisposition to the development of brain tumors. The most common of these conditions is neurofibromatosis type I, in which optic gliomas affect 5% to 15% of patients by age 20 years (3,4,5). In tuberous sclerosis, giant cell tumors also arise with significant frequency (6). Other genetic conditions that predispose to intracranial neoplasms include Gorland syndrome, basal cell nevus syndrome, Turcot syndrome (in which one-fourth of all cerebral neoplasms are PNETs), and tumors associated with neurofibromatosis type II (7).
The signs and symptoms of a CNS tumor with which an infant or child presents depend in part on the age of presentation but are often nonspecific relative to the type of tumor and relate more to its neuroanatomic location and secondary effects. Infants frequently present with an enlarging head circumference, nausea, and vomiting. Children may present with seizures or focal neurologic deficits such as hemiparesis. Tumors that develop in hormonally sensitive locations such as the hypothalamus may give evidence of disturbed endocrine function manifested by diabetes insipidus, growth failure, or precocious puberty. Tumors that involve the visual pathways often present as visual problems or impaired ocular motility.
Imaging Evaluation for Pediatric Central Nervous System Tumors
Although MRI is universally recognized as the most sensitive technique available, CT is very often the initial technique used because of its availability and the rapidity of the examination. With the current generation of CT scanners, multislice studies (64 to 128 slices) often take seconds, providing thin sections and relatively high resolution. The study of a brain tumor patient should include both pre- and postcontrast enhancement to determine whether hemorrhage, calcification, or contrast-enhancing tumor is present. If an MRI is scheduled to follow, then a pre-enhancement CT only may suffice. It should be understood that MRI is neither sensitive nor specific for calcification, so the only reliable way to detect calcification is by CT scanning. Although the CT scan may be adequate for the diagnosis and localization of a tumor mass, it may not be sufficient for characterization of the tumor type, for delineation of the tumor and its spread to the degree that can be done with MRI. CT scans are considered as a precursor to the MRI examination, if they are used at all.
The basic, routine MRI for the evaluation of the pediatric brain tumor depends in part on tumor location and size. In case of tumors of suprasellar and pituitary regions, there is a necessity for thin sections. In general, a sagittal and axial T1, axial and coronal T2, and axial and coronal fluid-attenuated inversion recovery (FLAIR) are performed prior to the injection of gadolinium contrast material. If clinically indicated, based on the suspicion of hemorrhage or calcification being present, a T2 gradient-echo susceptibility or a susceptibility-weighted imaging (SWI) scan can be performed before the injection of contrast material. Following the injection of contrast material, all three planes are obtained with T1 weighting. At the Children’s Hospital of Philadelphia, we favor the use in at least one plane (usually the axial) of fat saturation with T1-weighted imaging after the injection of contrast material. At the lower field strength of 1.5 Tesla (T), we use magnetization transfer sequences to increase the sensitivity to enhancement on our postcontrast images. At 3 T, we do not use magnetization transfer but do use fat saturation in at least one plane. Prior to the contrast injection we routinely perform arterial spin labelling (ASL) for assessment of cerebral blood flow, which is increased in the more vascular and more malignant tumors.
Diffusion imaging is included on all evaluations of the CNS, including all cases of brain tumors. The diffusion imaging is
performed as the last part of the examination because the pulse sequence tends to have a louder noise level and may awaken the sedated child. Depending on the neurosurgical approach to brain tumors at the institution where the child is being studied, navigational images may be acquired at the time of the initial MRI examination or later, prior to surgery. The navigational study includes axial postcontrast-enhanced T1 and T2 1-mm-thick slices obtained through the entire head at zero-degree angulation to the orbitomeatal line. The neurosurgeon can use these to plan the surgical approach and define the least hazardous pathway prior to surgery. Additional procedures that may be employed in the evaluation of the pediatric brain tumor by MRI include proton spectroscopy and task activation functional MRI when the tumor may be in or near a sensitive region, such as speech or language area, visual center, or motor cortex. These additional studies provide information about the risks involved and aid in planning the operative approach. Diffusion tensor imaging with fiber tracking can provide specific information about the effect of the tumor on tracts, whether they are displaced or infiltrated. Thus, diffusion tensor imaging with fiber tracking is a technique that has a potential in identifying vital pathways that may be at risk of injury or interruption by the surgical procedure. Once again, because most of these advanced MRI techniques are generally signal-to-noise starved, it is advantageous to use higher-field 3-T scanners for complete evaluation.
performed as the last part of the examination because the pulse sequence tends to have a louder noise level and may awaken the sedated child. Depending on the neurosurgical approach to brain tumors at the institution where the child is being studied, navigational images may be acquired at the time of the initial MRI examination or later, prior to surgery. The navigational study includes axial postcontrast-enhanced T1 and T2 1-mm-thick slices obtained through the entire head at zero-degree angulation to the orbitomeatal line. The neurosurgeon can use these to plan the surgical approach and define the least hazardous pathway prior to surgery. Additional procedures that may be employed in the evaluation of the pediatric brain tumor by MRI include proton spectroscopy and task activation functional MRI when the tumor may be in or near a sensitive region, such as speech or language area, visual center, or motor cortex. These additional studies provide information about the risks involved and aid in planning the operative approach. Diffusion tensor imaging with fiber tracking can provide specific information about the effect of the tumor on tracts, whether they are displaced or infiltrated. Thus, diffusion tensor imaging with fiber tracking is a technique that has a potential in identifying vital pathways that may be at risk of injury or interruption by the surgical procedure. Once again, because most of these advanced MRI techniques are generally signal-to-noise starved, it is advantageous to use higher-field 3-T scanners for complete evaluation.
Infratentorial Tumors
Unlike in the adult population, where infratentorial tumors typically consist of extraaxial tumors like acoustic schwannomas and meningiomas and intraaxial tumors are mostly metastases, in the pediatric age group, infratentorial tumors represent primary neoplasms of the cerebellum and brainstem. It is only in the adolescent that extraaxial tumors, such as acoustic schwannomas and meningiomas, begin to be encountered, and then primarily in the situation of neurofibromatosis type II. Disseminated tumors of the CNS, originating from either infratentorial or supratentorial primary tumors, can seed the subarachnoid space and grow onto the surface of the cerebellum and brainstem.
Cerebellar Tumors
The three most common pediatric brain tumors of the cerebellum are, in decreasing order of frequency, the PNET (medulloblastoma), cerebellar astrocytoma, and ependymoma (8). Other cerebellar neoplasms include the hemangioblastoma, oligodendroglioma, and lymphoma. All of these are relatively infrequent, with the hemangioblastoma occurring in the adolescent in association with von Hippel–Lindau syndrome. Cerebellar tumors can be divided into those that arise primarily within the cerebellar hemispheres and vermian tissue, the astrocytoma, and PNET, and those that arise primarily within the fourth ventricle, the ependymoma, and choroid plexus papilloma (CPP) or carcinoma.
Cerebellar Astrocytomas
Cerebellar astrocytoma is the most common cerebellar hemispheric tumor of glial origin, a tumor that is in the posterior fossa is second in frequency to the PNET. Cerebellar astrocytomas account for more than 10% of pediatric intracranial tumors and 25% of all posterior fossa tumors of children (9,10). There is no gender predilection. The most frequent astrocytoma is the pilocytic type. It should be noted that other glial-origin tumors arise in the cerebellar hemispheres, but less commonly. Patients with cerebellar astrocytomas usually present with symptoms of less than 2 months’ duration. Appendicular ataxia exceeds truncal and gait ataxia and precedes symptoms of increased intracranial pressure (8). Incidental astrocytomas may be found in the imaging studies of patients with predisposing conditions such as neurofibromatosis type 1 (NF1) or when imaging is done for conditions such as head injuries when the patient has no symptoms related to the posterior fossa. The pilocytic astrocytoma (PA) falls under the World Health Organization (WHO) classification as grade I, meaning extremely benign and generally curable if it can be completely resected. Fibrillary astrocytomas are the next most frequent glial tumors of the cerebellum. They represent approximately 15% of the glial tumors, compared to 85% for the pilocytic astrocytomas. These are more infiltrative and thus more difficult to totally resect and therefore to cure. Other less common glial neoplasms in the cerebellar hemispheres include ganglioglioma (GG) and oligodendroglioma. Glioblastomas of the cerebellum are very rare. They may be radiation induced and can be seen in patients who have been previously successfully treated for a brain tumor, such as PNET of the cerebellum. They tend to arise many years after the original therapy.
TABLE 9.1 Posterior Fossa Tumors in Childhood | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The juvenile pilocytic astrocytomas (JPAs), most commonly present between 5 and 15 years of age, are characterized by a slow growth rate, and only occasionally metastasize within the cerebrospinal fluid (CSF) pathways (Table 9.1). Although unencapsulated, they remain remarkably circumscribed and do not undergo malignant differentiation with time. Grossly, the tumors are cystic, with mural nodules in 50%, and more solid with various cystic central cavities in 40% (Fig. 9.1) (11). Completely solid tumors comprise only 10% of the total. Calcification is uncommon, reported to be at most, in 20% of JPAs. The JPA lacks a blood–brain barrier (BBB), and therefore fluid leaks and accumulates, first as microcysts around the tumor nodule or within the nodule, and eventually coalesces into macroscopic cysts (Fig. 9.2) (11). This explains the frequent cystic nature of the tumor, as well as the associated edema. The disturbance in the BBB results in the high incidence of contrast enhancement on both CT and MRI. Twenty-five-year survival rate for patients with JPAs is on the order of 90% or better (12).
Recurrence rates after gross total resection for cerebellar pilocytic astrocytomas vary from 0% to 12% (13). Based on their very slow growth rate, long-term progression-free survival may be seen even after subtotal resection. Second surgery for gross total resection is the method of choice for residual or recurrent low-grade cerebellar astrocytoma.
Recurrence rates after gross total resection for cerebellar pilocytic astrocytomas vary from 0% to 12% (13). Based on their very slow growth rate, long-term progression-free survival may be seen even after subtotal resection. Second surgery for gross total resection is the method of choice for residual or recurrent low-grade cerebellar astrocytoma.
 FIGURE 9.1 Cystic cerebellar pilocytic astrocytoma on gross specimen. (Courtesy of Dr. N. K. Gonatas, Philadelphia, PA.) |
 FIGURE 9.2 Pilocytic astrocytoma, microscopic section. Note the biphasic appearance, with both loose and dense areas of bipolar fusiform cells with bland nuclei in a fibrillary matrix. Small and large cystic formations are typical in this lesion. |
Computed Tomography
CT findings of cerebellar astrocytoma are those of a mass arising in the vermis or the cerebellar hemispheres, the solid portion being less dense than the surrounding brain, with both the solid and cystic portions typically being slightly denser than CSF (Fig. 9.3A). The density of
the cystic portion is due to the proteinaceous nature of the fluid. These tumors on CT are less dense than PNET and less often calcified than ependymomas. In more than 75% of the tumors, the solid portion of the mass shows enhancement. The solid portion may represent a large component of the astrocytoma or only a mural nodule, or it may be just a part of the tumor cyst wall (Fig. 9.4A,B). A cyst that is not lined by tumor should not enhance. The majority of the symptomatically presenting cerebellar astrocytomas have associated hydrocephalus due to compression or due to direct involvement of the fourth ventricle by the tumor. Fibrillary astrocytomas of the cerebellum are often less well defined than the pilocytic astrocytomas, with more irregular margins, and are more likely to have calcifications. Oligodendrogliomas of the cerebellum tend to be ill defined and densely calcified. Glioblastomas of the cerebellum are rapidly growing, irregularly enhancing, partially necrotic, infiltrative masses.
the cystic portion is due to the proteinaceous nature of the fluid. These tumors on CT are less dense than PNET and less often calcified than ependymomas. In more than 75% of the tumors, the solid portion of the mass shows enhancement. The solid portion may represent a large component of the astrocytoma or only a mural nodule, or it may be just a part of the tumor cyst wall (Fig. 9.4A,B). A cyst that is not lined by tumor should not enhance. The majority of the symptomatically presenting cerebellar astrocytomas have associated hydrocephalus due to compression or due to direct involvement of the fourth ventricle by the tumor. Fibrillary astrocytomas of the cerebellum are often less well defined than the pilocytic astrocytomas, with more irregular margins, and are more likely to have calcifications. Oligodendrogliomas of the cerebellum tend to be ill defined and densely calcified. Glioblastomas of the cerebellum are rapidly growing, irregularly enhancing, partially necrotic, infiltrative masses.
 FIGURE 9.3 Cystic cerebellar pilocytic astrocytoma. A 10-month-old boy with enlarging head circumference. A: Axial noncontrast-enhanced computed tomography shows hydrocephalus with cerebrospinal fluid reabsorption around frontal and temporal horns (arrowheads). This is due to a large mass in the left cerebellar hemisphere (arrows). The mass has a lower-density cystic component and a higher-density, more solid component (arrow). The fourth ventricle is displaced, and there is low-density edema in the brainstem and vermis. B: Axial enhanced T1 magnetic resonance imaging (MRI), fat saturated. There is intense enhancement of the solid components of the cystic tumor. C: Coronal enhanced T1 MRI postgadolinium, nonfat-saturated, showing tumor enhancement. |
 FIGURE 9.4 Solid cerebellar vermian astrocytoma. A: Axial noncontrast computed tomography (CT) shows a vermian mass projecting into the fourth ventricle. The mass is slightly less dense than the cerebellum. B: Axial contrast-enhanced CT. There is enhancement throughout most of the tumor. C: Axial proton density magnetic resonance imaging (MRI) shows high-signal-intensity mass. D: Axial T2 MRI also shows high signal in the mass. E: Precontrast material injection T1 MRI. The mass is less intense than the cerebellum but higher than CSF. F: Contrast-enhanced T1 MRI. The enhancement of the tumor is analogous to that shown on CT in panel B. |
Magnetic Resonance Imaging
Cerebellar astrocytomas are hypointense on T1 and hyperintense on T2, proton density, and FLAIR (Figs. 9.4C–E and 9.5A). The solid portion of the tumor is typically more hyperintense than the solid component of otherwise similarly appearing cystic PNET. Analogous to CT findings, the vast majority of the astrocytomas show contrast enhancement at the site of the solid tumor (Figs. 9.3B,C, 9.4F, and 9.5B). Diffusion studies show increased motion of water within both the cystic and the solid portions of the tumor, with increased apparent diffusion coefficient (ADC) values (Fig. 9.5C), another difference from the solid component of cystic PNET. Tumor dissemination remote to the site of the primary tumor is rare at the time of diagnosis or even in follow-up. Proton spectroscopy of low-grade cerebellar astrocytomas shows slight elevation of choline compared to N-acetyl aspartate (NAA), with a ratio of approximately 1.6:2.3:1 (14). This ratio is only from the solid portions of the tumor. The cystic portions of the tumor often show only elevated lactate and a lack of other metabolites. The presence of lactate in the cystic portion of the tumor has nothing to do with the tumor being aggressive, as in the case of malignant glioma, where it reflects anaerobic metabolism. Lactate in the cyst fluid of low-grade astrocytoma reflects the byproducts of metabolism that seep into the cysts. There is a low incidence of blood products in the wall of the cyst, and these manifest as marginal hypointensity on T2 and on susceptibility scans due to deposition of hemosiderin. Cerebellar astrocytomas do not present acutely as hemorrhagic masses, a finding that, when present, suggests a more aggressive tumor such as a PNET, atypical teratoid rhabdoid tumor (ATRT), or a tumor with a tendency to hemorrhage such as an ependymoma.
 FIGURE 9.5 Cystic pilocytic astrocytoma involving the vermis. A: Coronal fluid-attenuated inversion recovery magnetic resonance imaging (MRI). The solid component of the tumor has higher signal than the cystic component. B: Contrast-enhanced axial T1 MRI. There is enhancement of the solid component of the cystic tumor. C: Apparent diffusion coefficient (ADC) map MRI. The diffusion in the solid tumor is increased but not as much as within the tumor cyst. |
Postoperative evaluation of a patient with a cerebellar astrocytoma is generally performed within 24 hours of surgery. The reason for this is to avoid postoperative contrast enhancement at the site of reactive granulation tissue, which could be misinterpreted for residual tumor. Several days after surgery, granulation tissue starts to form, with disturbance in the BBB, and enhancement occurs at the margins of the resection whether there is or there is no residual tumor. In general, the most sensitive postoperative technique to detect residual tumor is MRI. CT is used primarily in the postoperative period for evaluation of residual hydrocephalus and/or other complications following surgery, such as the presence of extraaxial collections or intracerebral hematomas.
Whereas pilocytic astrocytomas of the cerebellum, most often cystic, are the most frequently encountered form of astrocytic tumor in the cerebellum, other forms do occur. Fibrillary astrocytomas are infiltrating and poorly defined tumors, and they may extend from the cerebellar peduncles into the brainstem; often they do not enhance. Their prognosis is less optimistic than that of the PA because over time the residual tumor can undergo malignant degeneration. GGs of the cerebellum do occur and are much less frequent than pilocytic astrocytomas. Like their counterpart in the supratentorial brain, GGs may be either cystic or solid, may contrast-enhance or not, and may be calcified. There is no easy way to differentiate these from other cerebellar astrocytic tumors except by pathology examination. Glioblastomas of the cerebellum are rare primary neoplasms. When they are found, they are more frequently the result of prior therapy, such as radiation therapy for a posterior fossa PNET that had been cured. Such a glioblastoma is detected during the long-term follow-up, often when the patient is a teenager or adult. More frequent than these glioblastomas of the cerebellum are radiation-induced dural-based meningiomas, which occur a decade or more after the completion of radiation therapy. When one meningioma is found postradiation and removed surgically, further follow-up imaging should be instituted because additional meningiomas are likely to develop with time.
In postoperative follow-up of any cerebellar tumor, if there is a finding that suggests residual tumor, a closer follow-up is needed. Scar tissue will not grow but remains stable or shrinks, whereas tumor tissue will grow with time. When recurrence is detected, the feasibility of re-resection is contemplated. In the immediate postoperative period, comparison to preoperative imaging study will permit detection of residual tumor if there is significant amount. Under these circumstances, neurosurgeons are often willing to go back in at that time and resect the remaining tumor.
As more and more MRI is used to study infants, children, and adolescents with symptoms such as developmental delay and headaches or posttrauma, a number of incidental lesions are being found. In such circumstances, a relatively frequent finding is an area of abnormal increased signal on T2 and FLAIR in the cerebellum. First, changes secondary to NF1 should be excluded by checking other sites within the brain. The stigmata of NF1 include bilateral and multiple sites of high T2 signal in cerebellar dentate nuclei and brainstem and in supratentorial brain, particularly globus pallidus. If these are present, then spongiform white matter dysplasia of NF1 is the likely etiology of the focus of high signal in the cerebellum. If spongiform changes of NF1 have been excluded, then the likelihood is that a tumor of the cerebellum has been found incidentally. Further evaluation with proton spectroscopy to look for elevated choline and depressed NAA levels can be useful if the lesion is large enough (greater than 1 cm × 1 cm × 1 cm). Such incidentally found lesions are followed with repeat contrast-enhanced imaging every 6 to 9 months. Some of these enhance initially, or with time begin to contrast-enhance. If the lesion enlarges or begins to contrast-enhance, then it is surgically removed. The experience has been that the lesions that grow over time turn out to be neoplasms, most often low-grade astrocytomas. Without evidence of another disease process, such as NF1 or a demyelinating disease, such lesions should be suspected to represent neoplasm and should be systematically followed with imaging.
Primitive Neuroectodermal Tumors of the Posterior Fossa (Medulloblastoma)
PNETs can arise in different locations and have been known by different names depending on their location. The different sites of origin of the PNET include the posterior fossa, where the tumor has been called medulloblastoma; the pineal region, where it is called pineoblastoma; the supratentorial brain, where it is called central neuroblastoma (NB); and the eye, where it is referred to as retinoblastoma. These tumors are WHO grade IV (Fig. 9.6).
Posterior fossa PNET is the most common malignant pediatric brain tumor, accounting for 15% to 30% of all pediatric CNS tumors and 30% to 55% of posterior fossa tumors (8). Between 250 and 500 pediatric brain tumors are newly diagnosed each year in the United States (8). PNETs of the cerebellum and vermis have an incidence of 0.5 to 0.7 per 100,000 (8).
The posterior fossa PNETs are more frequent in Hawaiians and in Maori of New Zealand, where the incidence approaches 12 per million children. In non-Hawaiian children in the United States, the incidence of posterior fossa PNETs is 5 to 9 per million children. The peak age for presentation of these tumors is between 5 and 7 years, with 80% diagnosed between ages 1 and 10 years (15). The male-to-female ratio is from 1.3 to 2.7 to 1 (9,15). From 1% to 2% of PNETs occur in association with a genetic tumor syndrome (16). Four percent of patients with Gorlin syndrome develop PNETs (17,18), and in Turcot syndrome, 25% of all cerebral neoplasms are PNET.
The posterior fossa PNETs are more frequent in Hawaiians and in Maori of New Zealand, where the incidence approaches 12 per million children. In non-Hawaiian children in the United States, the incidence of posterior fossa PNETs is 5 to 9 per million children. The peak age for presentation of these tumors is between 5 and 7 years, with 80% diagnosed between ages 1 and 10 years (15). The male-to-female ratio is from 1.3 to 2.7 to 1 (9,15). From 1% to 2% of PNETs occur in association with a genetic tumor syndrome (16). Four percent of patients with Gorlin syndrome develop PNETs (17,18), and in Turcot syndrome, 25% of all cerebral neoplasms are PNET.
 FIGURE 9.6 Primitive neuroectodermal tumor with subarachnoid space dissemination. A: Note the densely cellular proliferation of sheets of cells with small dark nuclei and scarce cytoplasm, along with a high mitotic rate. There are occasional rosette formations of nuclei around an amorphous anuclear center. (Courtesy of Dr. N. K. Gonatas, Philadelphia, PA.) B: Sagittal T1-weighted image shows midline posterior fossa mass extending inferiorly, mimicking inferior vermis and cerebellar tonsil. Note the trapped fourth ventricle with herniation of the fourth ventricle upward through the tentorial notch. Axial T2-weighted (C) and fluid-attenuated inversion recovery (D) images confirm the presence of a midline mass dorsal to the fourth ventricle and demonstrate hydrocephalus and characteristic low-intensity reflecting hypercellularity of the tumor. E: Scattered irregular enhancement, as opposed to diffuse intense enhancement, is noted, which is rather typical of such hypercellular tumors, as in the category of the primitive neuroectodermal tumors. |
Eighty percent of the PNETs involve the vermian tissue to some extent (8). Twenty percent arise laterally in the cerebellar hemisphere without involvement of the vermis. It is thought that in the younger children, the vermis, the inferior medullar velum, is the primary source of origin, and in the adolescent and older patient, the superior cerebellar hemisphere is the site of origin. Tumors arising laterally in the adolescent and young adult tend to be more desmoplastic than those that arise in the vermis of the younger patient. Brainstem involvement is seen in up to 38% of patients, and the incidence of hydrocephalus in more than 90% in some series (19,20,21). Although cysts can be seen within the tumor from 20% to 80% of cases, they are less a feature of PNET and more typical of the cerebellar astrocytoma; in addition, in the PNET the cysts tend to be small. PNETs characteristically present with a brief period of symptoms. In one series, symptoms were present for less than 6 weeks in half of patients and less than 12 weeks in three-fourths of patients (15). The symptoms are due to hydrocephalus and consist of headache, irritability, vomiting, blurred vision, and frequently ataxia. In 10% or less, the neurologic picture is that of acute decompensation brought on by hemorrhage into the tumor, causing acute hydrocephalus or brainstem compression (8).
Recent advances in molecular subgrouping of medulloblastomas have revealed important implications (22). Tumor location and enhancement patterns prove to be important imaging markers of subtypes. Group 3 and 4 tumors are midline fourth ventricular. Group 4 tumors have minimal to no enhancement. Sonic hedgehog tumors arise in the cerebellar hemisphere. Wingless tumors (WNT) arise in the cerebellar peduncle/cerebellopontine angle. These WNT have a good prognosis, whereas Group 3 tumors have worse survival. Modern imaging, along with improved surgical radiation and chemotherapeutic techniques, has resulted in an improvement in 5-year survival rates for patients with this tumor, from 30% in the 1960s to current rates of 50% to 70% (23).
 FIGURE 9.7 Primitive neuroectodermal tumor (PNET) of the cerebellum in a 2-year-old boy. A: Axial nonenhanced computed tomography. The tumor mass (arrows) is of greater density than the brainstem. B: Axial T2 magnetic resonance imaging (MRI). The tumor contains numerous small cysts. The solid component is relatively hypointense to cerebellum. C: Axial fluid-attenuated inversion recovery MRI. The tumor is mostly isointense to cerebellum. D: Axial T1, contrast enhanced and fat saturated. The tumor enhances. E: Axial diffusion MRI. There is decreased diffusion within the tumor. |
Computed Tomography
CT can be an important diagnostic aid in characterization and differential diagnosis of PNET. On CT, the precontrast enhancement density is either isodense or slightly increased at the site of the solid portion of the tumor (Fig. 9.7A) (24). Cystic portions are hypodense. Contrast enhancement occurs in 95% of cases. Eighty percent arise in the midline, involving the vermis to some extent. Hydrocephalus is present in greater than 90% of patients presenting with this type of neoplasm. Rarely—approximately less than 5% of the time—the increased
density in the tumor may reflect an acute bleed. The typical precontrast increased density seen in the PNET of the posterior fossa reflects the high cellularity—the greater amount of nuclear material compared to relatively low cytoplasmic and extracellular content. Although grossly disseminated tumor can be identified on CT before and/or after the injection of contrast, CT is a poor modality for the detection of disseminated tumor when compared to the high sensitivity of contrast-enhanced MRI (Figs. 9.8B and 9.9).
density in the tumor may reflect an acute bleed. The typical precontrast increased density seen in the PNET of the posterior fossa reflects the high cellularity—the greater amount of nuclear material compared to relatively low cytoplasmic and extracellular content. Although grossly disseminated tumor can be identified on CT before and/or after the injection of contrast, CT is a poor modality for the detection of disseminated tumor when compared to the high sensitivity of contrast-enhanced MRI (Figs. 9.8B and 9.9).
 FIGURE 9.8 Primitive neuroectodermal tumor disseminated to the spinal subarachnoid space at diagnosis with subsequent diffuse intracranial spread. A: Sagittal enhanced, fat-saturated T1 magnetic resonance imaging shows tumor enhancement (arrowheads) along the dorsal aspect of the thoracic cord. B: Ten months later, axial unenhanced computed tomography shows diffuse subarachnoid tumor seeding, seen as areas of increase density (arrowheads). |
 FIGURE 9.9 PNET dissemination. PNET tumor in 9 year-old girl that is disseminated throughout the subarachnoid space intracranially and intraspinally. A: Coronal T1 post gadolinium shows tumor in fourth ventricle and subarachnoid spread. B: Diffusion-weighted images show restricted motion of water in primary tumor as well as disseminated tumor. C,D,E: T1-weighted images of spine show disseminated tumor coating the spinal cord. |
Magnetic Resonance Imaging
A review of MRI findings in 42 consecutive Children’s Hospital of Philadelphia patients with posterior fossa PNETs revealed the tumors to be hypointense on T1 in 100%, isointense to the cerebellar folia on T2 in 95%, and contrast enhancing in 95% (unpublished data). There was restricted diffusion in 95% (Figs. 9.6A–D and 9.7B–D). Although these tumors most often enhance intensely, they can also show irregular enhancement or even none at all (Fig. 9.6E). These two findings (low
signal of the solid portion on T2 and restricted diffusion) are distinguishing features from JPA when evaluating a macrocystic posterior fossa mass. In those cases showing only mild or no enhancement (Fig. 9.10), this distinction becomes obvious because intense enhancement is a hallmark of JPA. PNET tumors may also be found incidentally (Fig. 9.11). Heterogeneity within the tumor on MRI prior to injection of contrast material can be due to hemorrhage, cysts, or necrosis (Figs. 9.11 and 9.12). The tumors that showed restricted diffusion and thus were hyperintense on diffusion sequence were dark and decreased in signal intensity on the ADC map (Fig. 9.7E). Measurements of ADC values for solid components of PNETs (range 0.67 to 0.99, mean 0.83 × 10−3 mm2/s) are consistently lower than those for ependymomas (range 1.0 to 1.3, mean 1.23 × 10−3 mm2/s) and can be useful for differential diagnosis (26). Proton spectroscopy shows evidence of malignant neoplasm by demonstrating a significant increase of choline relative to NAA, often a ratio of 3 or 4 to 1 (14). Taurine, an amino sulfonic acid, is also elevated in PNETs and may be demonstrated with proton spectroscopy performed with a short echo time of 20 to 35 ms. Taurine concentration in 13 patients with PNETs ranged from 2.62 to 11.15 mmol/kg with a mean of 6.09 ± 2.24. The mean concentration in 16 non-PNET tumors was 0.76 ± 0.95 mmol/kg. Thus, taurine concentration is another marker for the differential diagnosis of PNET from other tumors (27).
signal of the solid portion on T2 and restricted diffusion) are distinguishing features from JPA when evaluating a macrocystic posterior fossa mass. In those cases showing only mild or no enhancement (Fig. 9.10), this distinction becomes obvious because intense enhancement is a hallmark of JPA. PNET tumors may also be found incidentally (Fig. 9.11). Heterogeneity within the tumor on MRI prior to injection of contrast material can be due to hemorrhage, cysts, or necrosis (Figs. 9.11 and 9.12). The tumors that showed restricted diffusion and thus were hyperintense on diffusion sequence were dark and decreased in signal intensity on the ADC map (Fig. 9.7E). Measurements of ADC values for solid components of PNETs (range 0.67 to 0.99, mean 0.83 × 10−3 mm2/s) are consistently lower than those for ependymomas (range 1.0 to 1.3, mean 1.23 × 10−3 mm2/s) and can be useful for differential diagnosis (26). Proton spectroscopy shows evidence of malignant neoplasm by demonstrating a significant increase of choline relative to NAA, often a ratio of 3 or 4 to 1 (14). Taurine, an amino sulfonic acid, is also elevated in PNETs and may be demonstrated with proton spectroscopy performed with a short echo time of 20 to 35 ms. Taurine concentration in 13 patients with PNETs ranged from 2.62 to 11.15 mmol/kg with a mean of 6.09 ± 2.24. The mean concentration in 16 non-PNET tumors was 0.76 ± 0.95 mmol/kg. Thus, taurine concentration is another marker for the differential diagnosis of PNET from other tumors (27).
 FIGURE 9.10 A 5-year-old girl with vomiting. A: Axial T2-weighted image demonstrates large, partially cystic posterior fossa mass. Note that the solid component is low signal, that is, isointense to gray matter. B: Less than striking enhancement is also seen after intravenous contrast. The signal on T2 and the enhancement are typical of primitive neuroectodermal tumor (PNET) and not consistent with juvenile pilocytic astrocytoma as the diagnosis (surgically proven PNET). |
 FIGURE 9.11 Incidental primitive neuroectodermal tumor of the cerebellum. A 6-month-old boy with large head examined for possible benign enlargement of subarachnoid spaces. A: Axial T2 magnetic resonance imaging (MRI) reveals a small isointense mass (arrow) along the left side of the fourth ventricle. B: Coronal enhanced T1 MRI shows enhancement of the mass (arrow). |
MRI is a very important technique in the preoperative evaluation of PNETs because it is the only technique that shows the full extent of the primary tumor in all three planes (axial, coronal, and sagittal planes) and demonstrates with high sensitivity whether there is tumor dissemination. Dissemination may be present at the time of diagnosis prior to surgical resection in the cranial subarachnoid space, in the spinal subarachnoid space, and not infrequently at both sites (Fig. 9.8A). In our experience, dissemination to the spinal subarachnoid space was present in 11% of cases at the time of the initial diagnosis of the posterior fossa PNET. In follow-up with a mean time of 2 years after surgical removal of the tumor, 22% of patients showed evidence of spinal dissemination (28). When there is tumor disseminated to the spinal subarachnoid space, there is significant morbidity and mortality, with 81% of patients dying within a median time of 10 months (Figs. 9.6 and 9.8) (28). Aggressive chemotherapy and radiation therapy have affected the survival rate but have not ultimately cured patients with disseminated disease. This brings up the question and controversy concerning surveillance neuroimaging: How effective is it, and how often and for how long should it be performed? The recommended interval between imaging studies is every 3 to 4 months during the first year and every 6 to 8 months in subsequent years, for a maximum of 7 to 8 years (23). Most medulloblastomas recur in close proximity to the primary tumor site with a mean time to recurrence of 13 to 15 months. The Great Ormand Street Hospital, London Sick Kids, and Toronto Sick Kids Hospital experiences have been that no child presents with recurrence of spinal disease alone without intracranial disease, and therefore their recommendation is only to do follow-up studies of the brain in those patients that did not have preoperative tumor dissemination or residual postoperative tumor (29).
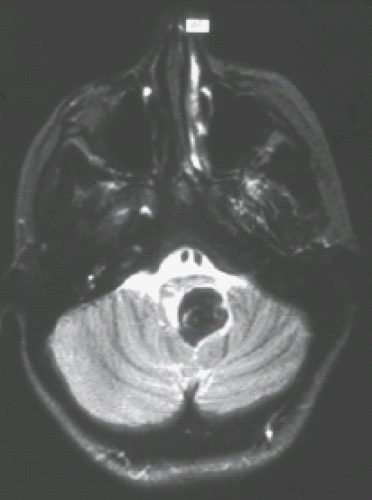 FIGURE 9.12 Hemorrhagic primitive neuroectodermal tumor of restiform body (inferior cerebellar peduncle) presenting as an acute bleed. T2 magnetic resonance imaging shows marked hypointensity of the acute bleed. Surgery revealed a primitive neuroectodermal tumor as the etiology of the hemorrhage. |
PNETs of the posterior fossa are divided into low risk and high risk based on patient’s age, tumor size, and metastatic stage. Low-risk tumors occur in patients older than 3 years, are up to 3 cm in the greatest dimension, and are without subarachnoid dissemination. High-risk tumors occur in patients younger than 3 years of age when radiation therapy is not feasible because of potential damaging effects of radiation therapy on the still-developing brain. These tumors are greater than 4 cm in the greatest dimension and have evidence of remote intraventricular or subarachnoid tumor dissemination. In case of low-risk tumors that have been completely resected and treated with both chemotherapy and radiation therapy, the 5-year survival rate is 75% to 80%.
As part of the initial workup of patients with PNETs, not only is the brain examined preoperatively, but so is the spinal canal. The imaging protocol that we use for the spinal canal includes sagittal T2, sagittal T1, and contrast-enhanced sagittal and axial T1 of the entire spinal canal. Obtaining just sagittal contrast-enhanced images is fraught with difficulty. The tumor adherent to the pial surface of the spinal cord or nerve roots is best seen in the axial projection, and there may only be a question of metastasis raised on the basis of the sagittal images. In addition, when the spinal canal is examined only postoperatively, there can be issues because of an extent of hemorrhage into the spinal canal from the surgery in the posterior fossa. The high T1 signal intensity of blood products may mimic disseminated tumor on an enhanced study. There can also be the postoperative development of subdural hygromas that extend from the posterior fossa down through the spinal canal and tend to accumulate contrast material within them, confusing the picture of whether there is spinal dissemination of tumor. Furthermore, in the region of the cauda equina, fatty lipomas of the filum terminale may mimic disseminated tumor on post-enhanced image, especially if no pre-enhanced study is available for comparison. In the follow-up of patients who have undergone surgery for posterior fossa PNET, the routine is to get immediate postoperative study to exclude any residual tumor. The subsequent follow-up is done as previously stated.
A particularly difficult issue is the evaluation of the non–contrast-enhancing PNET either in the brain or spinal canal, not only preoperatively, but also postoperatively, where less than gross dissemination of tumor may be very difficult to recognize (30). Techniques other than enhanced MRI may aid in recognition, such as relatively high-resolution diffusion imaging, looking for areas of restricted diffusion involving the subarachnoid space or ventricular lining. It remains to be seen whether newer targeted contrast agents or emerging molecular imaging methods can play a role in characterizing these tumors. Patients who survive long term are at risk of radiation-related injury (Fig. 9.13) and secondary neoplasms, such as meningiomas and glioblastoma multiforme (GBM).
 FIGURE 9.13 Radiation injury/necrosis. A 12 year-old girl many years post radiation therapy for PNET. A,B: Axial FLAIR images show abnormal hyperintensity at sites of white matter injury. C: T1 axial image post contrast shows bilateral enhancement of middle cerebellar peduncles. |
Atypical Teratoid/Rhabdoid Tumor
Atypical teratoid/rhabdoid tumor is a highly malignant tumor that arises not only in the CNS but also in the kidney and other organs. It occurs at a younger age than PNET and is categorized also as a WHO grade IV tumor, but it is more malignant, and if it originates in the CNS, it carries a very poor prognosis; survival is rare (30,31,32,33). The appearance on both CT and MRI is similar to that of PNET (Fig. 9.14) (34). In fact, histologically, there often are large areas of PNET within the ATRT tumor (31). However, in addition, there are areas of rhabdoid cells that appear quite different from the PNET component. ATRTs comprise 1.3% of pediatric CNS tumors (35). They comprise 6.7%
of CNS tumors in patients less than 2 years of age (34). By location, 35% to 65% are in the infratentorial region, 27% to 62% supratentorial, and 4% to 8% are both supra- and infratentorial in origin, that is, multifocal (30,31,32). Dissemination at diagnosis is present in between 15% and 34% of cases (31,32). One year after treatment, an additional 35% of tumors have disseminated (34). MRI studies positive for tumor dissemination have a concurrent incidence of positive cytology on CSF examination in just over half (56%) of cases (34). The survival rate at 1 year is 71% and at 5 years is 28% (34). Clues that the tumor is of ATRT type are clinical, with the age at the onset of the tumor being very young. These tumors also show a tendency to occur in the cerebellar pontine angle or medullary pontine angle, appearing like an extraaxial mass, not infrequently hemorrhaging and projecting into the foramen magnum.
of CNS tumors in patients less than 2 years of age (34). By location, 35% to 65% are in the infratentorial region, 27% to 62% supratentorial, and 4% to 8% are both supra- and infratentorial in origin, that is, multifocal (30,31,32). Dissemination at diagnosis is present in between 15% and 34% of cases (31,32). One year after treatment, an additional 35% of tumors have disseminated (34). MRI studies positive for tumor dissemination have a concurrent incidence of positive cytology on CSF examination in just over half (56%) of cases (34). The survival rate at 1 year is 71% and at 5 years is 28% (34). Clues that the tumor is of ATRT type are clinical, with the age at the onset of the tumor being very young. These tumors also show a tendency to occur in the cerebellar pontine angle or medullary pontine angle, appearing like an extraaxial mass, not infrequently hemorrhaging and projecting into the foramen magnum.
 FIGURE 9.14 Posterior fossa atypical teratoid/rhabdoid tumor in a 10-month-old boy. A: Axial fluid-attenuated inversion recovery magnetic resonance imaging (MRI). Tumor mass is isointense to cerebellum, with higher signal surrounding edema. B: Axial enhanced T1 MRI. There is irregular enhancement of the tumor. |
Ependymomas of the Posterior Fossa
Posterior fossa ependymomas represent from 10% to 20% of pediatric posterior fossa tumors, or up to 8% of all pediatric brain tumors and 1.7% of all childhood cancers. The peak age incidence is between 3 and 6 years, with a second peak occurring in the middle years of adult life (36). The tumor arises within the fourth ventricle, extending into and through the outlets and the foramina of Luschka and the foramen Magendie, and giving impression of a plastic tumor mass (Fig. 9.15). Ependymomas are well-differentiated, moderate cellular gliomas with perivascular pseudorosettes, rare mitoses, and occasional areas of necrosis, hemorrhage, and calcification (Fig. 9.16). They are classified as WHO grade II.
 FIGURE 9.15 Ependymoma fourth ventricle. A 11 month-old girl. A: Axial T2-weighted image shows a mixed signal intensity mass in the lateral recess of the fourth ventricle on the left. B: Susceptibility-weighted image (SWI) shows evidence of bleeding within the tumor mass. C: Postcontrast T1-weighted axial image shows inhomogeneous contrast enhancement. |
The overall incidence of ependymomas is 2.2 per million per year (20,21,37). Younger than the age of 5 years the incidence is 3.9 per million, and older than the age of 5 years the incidence is 1.1 per million. Forty percent of the cases occur younger than the age of 4 years, and 80% occur younger than the age of 8 years (38,39). There is no gender bias. Two-thirds of the cases occur in the posterior fossa and one-third in the supratentorial brain. This is a tumor that is not usually disseminated at the time of diagnosis, the incidence being approximately 5% (38,40). In general, local recurrence occurs, in 90% or more of the cases, before the tumor disseminates (41). The median time between tumor diagnosis and recurrence is 22 months (8). Dissemination occurring after initial diagnosis and before local recurrence is unusual, only 7% to 8% of cases (42). However, once the tumor has recurred locally and continues to grow, then dissemination becomes more likely, reaching 24% of cases. Dissemination often leads to death. In a series of 52 children with ependymoma, recurrences developed in 54%, with a median time from surgery to first recurrence of 14.5 months. Forty-three percent of recurrences were asymptomatic, having been picked up on surveillance imaging (43). Thus, surveillance imaging is recommended after the immediate postoperative
study for residual disease every 3 to 6 months for the first year and every 6 months for the next 4 years (43). If tumor recurs, then spinal imaging should also be performed.
study for residual disease every 3 to 6 months for the first year and every 6 months for the next 4 years (43). If tumor recurs, then spinal imaging should also be performed.
 FIGURE 9.16 Ependymoma, microscopic features. A: Histologic specimen shows cells with small bland nuclei and fibrillary eosinophilic matrix. B: Nuclear free areas, especially in perivascular regions, and tapering of fibrillary processes around blood vessels are characteristic features of ependymomas. |
Poor prognostic factors include patient age younger than 2 years, a short history of symptoms prior to presentation, presence of brainstem and cranial nerve deficits, lateral location of tumor, and high Ki-67 immunolabeling index (43).
The 5-year progression-free survival for ependymomas is 36% to 64% (44). The 10-year progression-free survival is only 47% to 48% (44). In the posterior fossa, the most favorable circumstance for an ependymoma is one that is still localized within the fourth ventricle, not extending through the outlets, and not involving the cranial nerves or blood vessels that lie lateral and anterior to the brainstem. A completely resected ependymoma of the fourth ventricle stands a good chance of cure on the basis of surgery alone. Radiation and chemotherapy, although commonly used, are not by themselves curative. The 10-year actuarial local control for gross total resection and radiotherapy was 100%, for just gross total resection it was 50%, and for subtotal resection and radiotherapy it was only 36%.
Computed Tomography
Ependymomas are known for the production of calcification and for a tendency to bleed. Thus, with both CT and MRI, there is often an inhomogeneity to the tumor before and after contrast material injection. On a pre-enhanced CT, the appearance of an ependymoma is that of a mass of mixed density (45,46,47,48). The mixed higher densities seen within the tumor can represent calcification or hemorrhage (Fig. 9.17). It is the localization within the fourth ventricle and the mixed densities that characterize the ependymoma. Contrast enhancement on CT is often inhomogeneous (Fig. 9.18) (45,46,47,48). Hydrocephalus is frequently present and depends on obstruction of the fourth ventricle or aqueduct of Sylvius. Extension of tumor through the outlets of the fourth ventricle into the cerebellopontine angle, in front of the brainstem, or down along the dorsal aspect of the cervical spinal cord may be visible on CT but is better seen on MRI.
 FIGURE 9.17 Ependymoma of the fourth ventricle. Axial noncontrast computed tomography (CT) shows a mass in the fourth ventricle containing calcifications producing hydrocephalus. |
 FIGURE 9.18 Ependymoma of the fourth ventricle. Contrast-enhanced computed tomography shows intrafourth ventricular mass with irregular contrast enhancement. |
Magnetic Resonance Imaging
The MRI appearance of the ependymoma is that of mixed T1 signal intensity, the high signal intensity not infrequently being the result of blood products or calcium within the tumor (48,49,50). On MR images the ependymoma is seen filling and distending the fourth ventricle (Fig. 9.15) and extending through the outlets (Figs. 9.18 and 9.19). On T2-weighted images, the intensities again are often quite variable, consisting of both increased and decreased signals (Figs. 9.15 and 9.20A). Again, it is the mixture of calcifications, blood products, and tumor
that contributes to this inhomogeneity. Contrast enhancement is also typically inhomogeneous (Figs. 9.15 and 9.20B). Critical to the preoperative evaluation is the delineation of the extent of the tumor, determining how far the tumor goes outside the fourth ventricle, whether it extends lateral and/or ventral to the brainstem, and whether it projects inferiorly along the cervical spinal cord. Any tumor that remains postoperatively will regrow and potentially can lead to death.
that contributes to this inhomogeneity. Contrast enhancement is also typically inhomogeneous (Figs. 9.15 and 9.20B). Critical to the preoperative evaluation is the delineation of the extent of the tumor, determining how far the tumor goes outside the fourth ventricle, whether it extends lateral and/or ventral to the brainstem, and whether it projects inferiorly along the cervical spinal cord. Any tumor that remains postoperatively will regrow and potentially can lead to death.
 FIGURE 9.19 “Plastic” ependymomas in two patients. A: Sagittal T1-weighted magnetic resonance imaging (MRI) (600/20). B: Sagittal T1-weighted MRI (600/20) with contrast enhancement. Masses (arrows) extend downward through the foramen magnum and mimic tonsillar herniation. Note the extensive enhancement of the second patient’s tumor after gadolinium (B). C: Axial T2-weighted MRI (6,000/99) shows a fourth ventricular mass extending through the foramina of Luschka bilaterally (arrows). |
As part of the preoperative MRI evaluation, both the brain and the spinal canal are examined, but the yield in the spinal canal is limited. In the postoperative follow-up of tumor, both CT and MRI play an important role in searching for residual tumor, which may be seen as calcification on CT (Fig. 9.21A) or as enhancement on MRI (Fig. 9.21B). Residual tumor, whether local or distant from the original site of surgery, may initially be difficult to detect. Small areas of tumor seeding from an ependymoma to the ependymal surface of the ventricular system, such as the lateral ventricles, often will not show contrast enhancement in the early stages of growth. Only when the tumor develops a larger blood supply will start to enhance (Fig. 9.22). A tumor that persists and grows over time eventually will seed to the subarachnoid space not only intracranially, but also in the spinal canal. As these tumors grow, they often become hemorrhagic.
Proton spectroscopy is a useful adjunct to the imaging studies because ependymomas have a different spectrum than do astrocytomas and PNETs, with preservation of creatine, elevation of choline, and decrease in NAA (14). The creatine is not preserved in PNET and astrocytoma.
 FIGURE 9.20 Ependymoma of the fourth ventricle in a 4-year-old boy. A: Axial T2 magnetic resonance imaging shows a heterogeneous hyperintense mass extending into the left Luschka foramen (arrow). B: Axial T1 postgadolinium image shows irregular enhancement. |
Choroid Plexus Papilloma
Choroid plexus tumors comprise 2% to 4% of pediatric brain tumors (35), and 10% to 20% of those that arise in the first year of life (35,51). For every six choroid plexus tumors, one is a carcinoma and five are papillomas. At the time of presentation, 80% occur in patients less than 2 years of age (52). Overall, there is equal distribution between males and females. Forty percent arise in the fourth ventricle, with a slight male predominance, 3:2. Our experience has been that in the fourth ventricle, the tumors have been CPPs and not carcinomas. Choroid plexus carcinomas (CPCs) are more common in the lateral ventricles. In the posterior fossa, the choroid plexus tumor produces obstruction leading to hydrocephalus. With both
supra- and infratentorial tumors, excess production of CSF is possible, resulting in hydrocephalus as well (53). Bleeding from the tumor into the ventricle can also lead to hydrocephalus by causing adhesions and obstruction of the CSF pathways. CPP of the fourth ventricle is most often cured by surgical procedure. Any benign tumor, if disrupted during the surgical procedure, can disseminate into the spinal canal or into the cerebral subarachnoid space.
supra- and infratentorial tumors, excess production of CSF is possible, resulting in hydrocephalus as well (53). Bleeding from the tumor into the ventricle can also lead to hydrocephalus by causing adhesions and obstruction of the CSF pathways. CPP of the fourth ventricle is most often cured by surgical procedure. Any benign tumor, if disrupted during the surgical procedure, can disseminate into the spinal canal or into the cerebral subarachnoid space.
 FIGURE 9.21 Residual ependymoma in two patients. A: A 2-year-old girl. Unenhanced computed tomography reveals calcification in the residual tumor. B: A 7-year-old boy. Enhanced T1 magnetic resonance imaging shows a contrast-enhancing tumor (arrow) in the left cerebellopontine angle. |
Computed Tomography
CPP of the fourth ventricle lies within the fourth ventricle and usually does not extend through the foramina into the adjacent subarachnoid space, as is the case with the ependymoma. The tumor is usually of uniform increased density on CT and enhances intensely and homogeneously (Fig. 9.23A) (54). The size is variable, from quite small—not much larger than the choroid plexus itself—to one that completely fills and distends the fourth ventricle.
Magnetic Resonance Imaging
The CPP typically on T1 is a hypointense mass, and it is usually somewhat hyperintense on T2 with focal hypointensities within it reflecting some of the blood flow that is quite rich within this tumor (Fig. 9.23B,C) (55). The enhancement pattern is typically homogeneous and bright (Fig. 9.23D). Often, the appearance of the tumor has a characteristic cluster-of-grapes morphology (Fig. 9.24) that resembles the overall appearance of native choroid plexus. After a negative postoperative study, there is usually no further need for significant follow-up and no need to examine the spinal canal.
 FIGURE 9.22 Ependymoma dissemination into the lateral ventricle, early and late changes. A: Axial enhanced T1 magnetic resonance imaging (MRI). A nonenhancing nodule (arrow) is noted adhering to the wall of the left lateral ventricle. B: One year later, coronal enhanced T1 MRI shows multiple enhancing nodules of disseminated tumor. |
Brainstem Gliomas
The exact incidence of tumors arising within the brainstem is not known because the sensitivity of MR in detection of small tumors, particularly in the midbrain, is unfolding. Overall incidence is 10% to 20% of all pediatric brain
tumors (56,61). Most of the literature has been devoted to the classic pontine glioma, a tumor that occurs most often between 5 and 6 years of age, is diffusely infiltrating, and tends to run a malignant course with a life expectancy not exceeding 14 months (57). Chemotherapy has not shown any role in increasing survival, and radiation therapy, the preferred method of treatment for diffuse pontine gliomas, extends life only by several months (57,58). The prognosis of a brainstem tumor depends on its site of origin, whether it arises in the midbrain, pons, and medulla, and to a lesser extent on how it appears (59,60).
tumors (56,61). Most of the literature has been devoted to the classic pontine glioma, a tumor that occurs most often between 5 and 6 years of age, is diffusely infiltrating, and tends to run a malignant course with a life expectancy not exceeding 14 months (57). Chemotherapy has not shown any role in increasing survival, and radiation therapy, the preferred method of treatment for diffuse pontine gliomas, extends life only by several months (57,58). The prognosis of a brainstem tumor depends on its site of origin, whether it arises in the midbrain, pons, and medulla, and to a lesser extent on how it appears (59,60).
 FIGURE 9.23 Choroid plexus papilloma of the fourth ventricle in two patients. A: Axial unenhanced computed tomography shows a small dense mass (arrow) in the inferior fourth ventricle. B: Sagittal T1 magnetic resonance imaging (MRI), hypointense fourth ventricular mass. C: On coronal T2 MRI, a hyperintense mass occupies the fourth ventricle. D: Sagittal enhanced T1 MRI reveals intense enhancement of the mass. |
 FIGURE 9.24 A 4-year-old girl with headaches. Sagittal T1-weighted (A) and axial T2-weighted (B) images show characteristic lobulated morphology of an intraventricular choroid plexus papilloma within the third and lateral ventricles. Note the marked enhancement after intravenous contrast on axial postcontrast T1-weighted images (C). |
 FIGURE 9.25 Pilocytic astrocytoma of the pons in a 17-year-old girl. A: Axial T2 magnetic resonance imaging (MRI) shows a left-sided, well-demarcated, high-signal-intensity mass in the pons. B: Sagittal enhanced T1 MRI shows diffuse contrast enhancement of the mass. C: Apparent diffusion coefficient (ADC) map MRI shows increased diffusion within the tumor mass. |
A recent multicenter clinicopathologic reappraisal of brainstem tumors divided the tumors into fibrillary astrocytomas, which involve the ventral pons and present with abducens palsy and have a grim prognosis, and focal brainstem gliomas, often pilocytic astrocytomas, which arise outside of the ventral pons and have a tendency to exophytic growth, long clinical prodrome, and good survival (Fig. 9.25). The prognosis is clearly influenced by any underlying predisposing disease state, for example, NF1, in which the pontine gliomas have a much more benign and prolonged course than in non-NF1 patients.
In general, tumors arising in the midbrain have a tendency to be indolent and of low-grade astrocytic nature, for which the treatment is often just shunting of the hydrocephalus that is secondary to the aqueductal occlusion (Fig. 9.26) (59,60). The tumors are followed by imaging, and if the tumor shows enlargement over time, then therapy in the form of chemotherapy and/or radiation therapy may be undertaken. The main problem with the midbrain tumors is that they can be mistaken for aqueductal stenosis on CT, where they can be missed and not detected until an MRI is performed. In addition, there can be a problem in differentiating a tumor of the tectal plate from a pineal region tumor because both may occupy the same space. When in doubt, the appropriate markers need to be obtained to rule out a germinoma, or on occasion it may be necessary to obtain a biopsy to differentiate the two. For the most part, tumors of the midbrain can be diagnosed on the basis of their location and MRI appearance. A subset of these tumors can extend into the thalamus, and these tend to have a less benign course, mainly related to their thalamic component. Midbrain tumors may eventually grow to involve the pons or cerebellum through the superior cerebellar peduncles, findings that are less frequent.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


