Fig. 1
Schematic diagram of radiolabeling a peptide or protein via a bifunctional chelating agent (BCA)
Table 1
Therapeutic radionuclides
Radionuclide | Type of emission | Physical half-life | Ionic radius (pm) | Charge | Emax (MeV) | Mean tissue penetration (mm) | Production |
|---|---|---|---|---|---|---|---|
β-emitters | |||||||
67Cu | β−, γ | 2.6 d | 74 or 71 | +1 or +2 | 0.57 | 0.6 | Cyclotron |
90Y | β− | 2.7 d | 104 | +3 | 2.3 | 2.76 | Generator |
177Lu | β−, γ | 6.7 d | 100.1 | +3 | 0.50 | 0.28 | Reactor |
186Re | β−, γ | 3.7 d | 72 | +5 | 1.07 | 0.92 | Reactor |
188Re | β−, γ | 17.0 h | 72 | +5 | 2.1 | 2.43 | Generator |
α-emitters | |||||||
211At | α | 7.2 h | 76 | +7 | 5.87 | 0.04–0.1 | Reactor |
212Bi | α | 1.0 h | 117 | +3 | 6.09 | 0.04–0.1 | Reactor |
213Bi | α | 45.7 min | 117 | +3 | 5.87 | 0.04–0.1 | Reactor |
225Ac | α, β− | 10 d | 126 | +3 | 5.83 | 0.04–0.1 | Reactor |
Auger emitters | |||||||
111In | Auger, γ | 2.8 d | 94 | +3 | 0.025 | <100 nm | Cyclotron |
201Tl | Auger, γ | 73.1 h | 150 | +1 | – | <100 nm | Cyclotron |
193mPt | Auger, γ | 4.3 d | 62.5 | +4 | 0.010 | <100 nm | Cyclotron |
195mPt | Auger, γ | 4.0 d | 62.5 | +4 | 0.023 | <100 nm | Cyclotron |
Unlike radioiodine, almost all inorganic radionuclides should be chelated with a BCA before they are attached to a biologically active molecule, although in some cases the chelating agent itself is the targeting agent. The metallic radionuclides are somewhat diverse in terms of their coordination chemistries, and thus, various kinds of BCAs are required to label with many radionuclides. The selection of a suitable BCA for a specific radionuclide involves the consideration of numerous factors based on the intended use; for example, the coordination number of the metal ion, the match between the size of the chelate cavity and the ionic radius of the metal ion, oxidation state, the hardness or softness of the ion, the chelate charge or number of donor binding groups, and the rates of complex formation and dissociation (Packard et al. 1999).
Many types of therapeutic radionuclide chelating agents, such as acyclic and macrocyclic compounds, have been reported (Fig. 2). Bifunctional EDTA (ethylenediaminetetraacetic acid) derivatives were initially developed to chelate 111In and 90Y. However, stability problems resulted in the developments of bifunctional diethylenetriaminepentaacetic acid (DTPA) derivatives stimulated to develop new derivatives that were more suitable in terms of coordination number and complex stability (Meares and Wensel 1984; Brechbiel et al. 1986). For example, one of the ethylene groups in DTPA was modified with a cyclohexyl carbon chain to avoid uncertainties concerning the structures of radionuclide-DTPA complexes, and CHX-DTPA was produced, which was subsequently labeled with 90Y, 111In, 177Lu, or 213Bi, and formed stable complexes (Wu et al. 1997; Brechbiel and Gansow 1992). However, although successful results were achieved using DTPA derivatives, the overall stabilities of radionuclide-DTPA complexes were less than perfect, which introduces the risk of toxicities caused by released radionuclides (Carrasquillo et al. 1999).
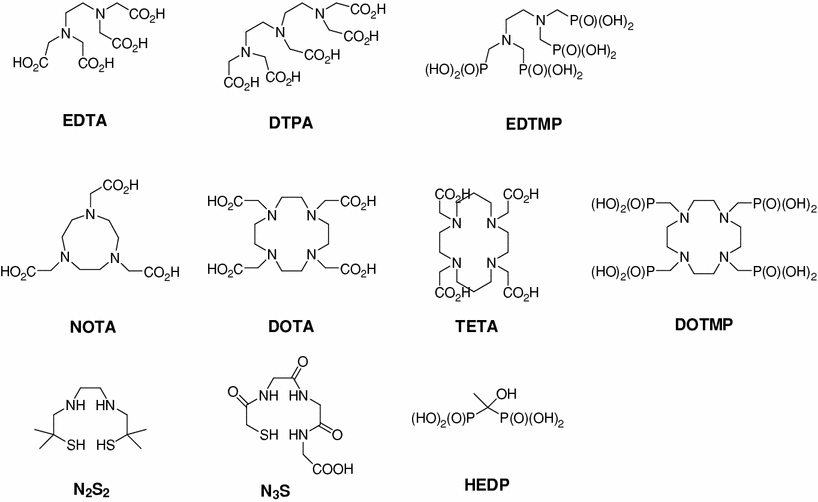
Fig. 2
Bifunctional chelating agents (BCA) commonly used for inorganic radionuclide labeling
Octadentate macrocyclic bifunctional 1,4,7,10-tetraazacyclododacane-1,4,7,10 -tetraacetic acid (DOTA) derivatives were developed for labeling with 90Y, 111In, and radio-lanthanides, and showed better stabilities than acyclic BCA complexes (Moi et al. 1988; Kline et al. 1991; Mcmurry et al. 1992; Eisenwiener et al. 2000). Nevertheless, although a large number of DOTA complexes have been developed for therapeutic purposes, they are limited by their slow complex formation rates, and thus, the requirement for a heating process for radiolabeling (Moreau et al. 2004; Li and Beheshti 2005).
Hexadentate macrocyclic bifunctional 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA) derivatives have also been produced for the labeling of In(III) (Clarke and Martell 1991), and form highly stable complexes with Ga(III). However, DOTA derivatives are better chelates of radio-lanthanides (Cacheris et al. 1987). Furthermore, the macrocyclic 14-membered ring, 1, 4, 8, 11-tetraazacyclotetradecane-1, 4, 8, 11-tetraacetic acid (TETA) has been reported to chelate Cu(II) (Cole et al. 1986; Moi et al. 1987; Cole et al. 1987).
The phosphorous-containing chelating agents, hydroxyethylidene diphosphonic acid (HEDP), ethylenediamine-N,N,N′,N′–tetrakis(methylenephosphonic acid) (EDTMP) and 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraaminomethylenephosphonic acid (DOTMP) have been reported to be useful for the radionuclide therapy of metastases to bone. HEDP has been used to complex 186Re and 188Re (Ketring 1987; Lin et al. 1997), and both EDTMP and DOTMP chelate 177Lu and 212Bi (Chakraborty et al. 2008; Hassfjell et al. 1997). Furthermore, diamine dithiol (N2S2) derivatives have been used to chelate 188Re, and have been utilized for the treatment of liver cancer (Jeong et al. 2001; Lee et al. 2002), and recently, an acetylated precursor of an N2S2 derivative was found to protect highly reactive sulfhydryl groups (Lee et al. 2007).
A BCA contains a radionuclide chelating region and a covalent linkage region, which is capable of conjugating a biologically active molecule with a radionuclide complex. A number of conjugation methods have been devised for conjugating biologically active molecules and BCAs, and have been applied to bioactive molecules, such as small molecules, peptides, or proteins containing amino or thiol groups (Fig. 3) (Fichna and Janecka 2003). The conjugation between ester, isothiocyanate, maleimide, or alkyne groups of a BCA and the amino, thiol, or azide groups of biologically active molecule provides representative examples. The selection of a conjugation method depends on the feasibility of the required chemical modification.
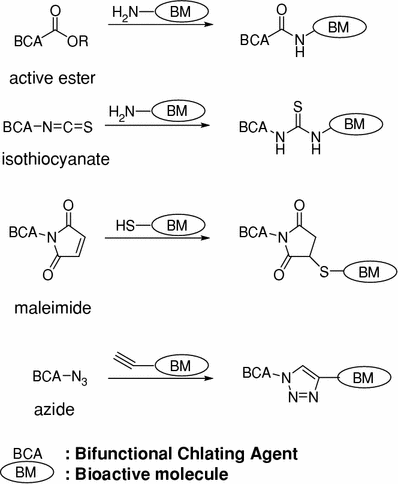
Fig. 3
Methods generally used for conjugating BCA and bioactive molecules
Here, we classify inorganic radionuclides as β- or α-emitters, or Auger electron emitters, and discuss the BCA-related chemistries and applications of several radionuclides.
3.1 β-Emitters
Beta particle emitters are the most extensively used for in vivo radionuclide therapy. In particular, a β-emitter, 131I has been used for therapy of thyroid cancer for over 70 years. β-particles are high energy electrons emitted from the nucleus at a continuum of energies. Their low ionization densities along their tracts is indicative of their poor LET abilities (Cobb and Humm 1986). In practice, this means that β-particles kill cells via indirect effects. β-particles have limited ability to irreversibly damage DNA, but instead, ionize neighboring molecules, especially water, to toxic radicals, peroxides, and oxides, which are capable of damaging DNA. Furthermore, this generation of toxic species is accelerated by temperature and the presence of oxygen. In addition, the high-energy β-particles emitted by these radionuclides have much greater mean tissue penetration depths than α-emitters or Auger electrons, and thus, high-energy β-particles also kill neighboring tumor cells by the so-called cross-fire effect (Schubiger et al. 1996; Howell et al. 1989).
β-emitters are mainly produced in nuclear reactors, which use the (n,γ) reaction to produce neutron-rich isotopes; the others are produced in charged-particle accelerators. However, most radionuclides produced in nuclear reactors have low specific activities. Radionuclides with high-specific activities can be obtained using generator systems, indirect production schemes, and sometimes by direct (n,γ)-activation (Volkert et al. 1991; Knapp FF Jr 1998). It is essential that only nuclear reactions and separation schemes that result in high purity products be used.
3.1.1 67Cu
67Cu (t1/2: 2.6 days) emits beta particles with maximum energies of 577, 484, and 395 keV (mean 141 keV), which is similar to the mean β energy of 131I (180 keV). These therapeutic β-emissions, with a mean tissue penetration depth of 0.2 mm, are suitable for treating small tumors of up to 5 mm in diameter. 67Cu also emits γ-radiation in the range 91–184 keV (49 % abundance), which is particularly suitable for diagnostic imaging with a gamma camera (Novak-Hofer and Schubiger 2002). 67Cu can be produced by bombarding 67Zn with neutrons in high flux reactors (Mausner et al. 1998), or in cyclotrons by irradiating natZn or enriched 68Zn with protons (Schwarzbach et al. 1995; Dasgupta et al. 1991).
67Cu has a formal charge of +2 in its complexed form, and the ionic radius of 67Cu2+ is 71 pm, and is commonly complexed with a macrocyclic BCA, such as TETA (Fig. 2) (Meares et al. 1984; Moi et al. 1985). 67Cu(II) can form stable octahedral complexes of coordination number 6 with DOTA and TETA. However, despite the high stability of the TETA-Cu complex, transchelation of Cu from the complex in liver has been observed in an in vivo experiment using a 67Cu-TETA-antibody (Mirick et al. 1999). Recently, cross-bridged tetraamine macrocyclic (cyclam) ligands with nonadjacent nitrogens connected by an ethylene bridge were synthesized. These ligands increase the reduction potential of Cu(II), and thus, retard its electrochemical reduction to Cu(I), which is believed to facilitate copper dissociation from these complexes (Boswell et al. 2004). However, the complexation of Cu(II) with TETA derivatives requires an additional linkage site to maintain bifunctionality, because TETA has 6 binding sites, which are used for coordination with Cu(II). Moreover, the chelation reaction requires an elevated temperature (75 °C) which is not optimal for the post-conjugation labeling of proteins (Novak-Hofer and Schubiger 2002).
67Cu is usually chelated by incubating it with BCA-conjugated peptides or proteins for 20–30 min at ambient temperature and pH ~5; excess and nonspecifically bound Cu(II) is sequestered with EDTA (Mirick et al. 1999).
67Cu has been used for radioimmunotherapy by conjugating it to monoclonal antibodies (mAbs) or antibody fragments, which bind to tumor-specific antigens expressed by a large number of tumors (Postema et al. 2001; Smith et al. 1993; Hughes et al. 1997; O’Donnell et al. 1999). 67Cu labeled receptor-specific peptide is also suitable for peptide receptor radionuclide therapy (PRRT). For example, 90Y- and 177Lu-octreotide derivatives have been used for treatment of neuroendocrine tumors. However, to date, radioactive copper-labeled peptides have only been used for 64Cu imaging studies (Anderson et al. 2001; Lewis et al. 1999; Prasanphanich et al. 2007).
3.1.2 90Y
90Y (β–, Emax = 2.3 MeV, t1/2 = 64 h) is of particular interest due to its high-energy pure β-particle emission with a 2.8 mm mean tissue penetrating depth. 90Y can be produced from a generator by the decay of 90Sr, and decays to stable 90Zr. 90Y has a half-life of 2.7 days, which is short enough to achieve a critical dosing rate and at the same time is long enough to allow the radiopharmaceutical to be manufactured and delivered for clinical use (Liu 2004). 90Y has a charge of +3 when complexed, and an ionic radius of 104 pm. The BCAs commonly used to complex 90Y are DTPA and DOTA derivatives (Brechbiel 2008).
The in vivo dissociation and transchelation reactions of 90Y are the main sources of its toxicity. For example, reactions between 90Y chelates and biologically important metal ions, such as Ca2+ and Fe3+, produce free 90Y, which concentrates in bone and causes bone marrow toxicity. Competition between BCA and native chelators, such as amino acids and transferrin, may also result in the early release of 90Y from 90Y-labeled BCA-bioactive molecule (BM) conjugates (Liu 2004). To resolve these problems, DOTA derivatives, which form highly stable complexes, are better choices for 90Y than DTPA derivatives (Carrasquillo et al. 1999; Eisenwiener et al. 2000).
To determine the biodistribution and radiation dosimetry of 90Y, 111In(III) is often used as a surrogate, because 90Y does not emit γ-ray which is required for scintigraphic imaging in humans. The chelation chemistry of 111In with polyaminocarboxylate ligands is similar to that of 90Y (Wilder et al. 1996; Gordon et al. 2002).
90Y is one of the most frequently used radioactive metals in clinical practice, due to its high-energy pure β emission and availability as a sterile pyrogen-free product with high specific activity. Radioimmunotherapy, which was initiated using 131I labeled monoclonal antibodies more than 30 years ago, provides a typical example of 90Y radionuclide therapy. After much research and several clinical trials, 90Y/anti-CD20 monoclonal antibody-based radioimmunotherapy was recently approved by the US Food and Drug Administration (FDA) as a therapeutic modality for B cell non-Hodgkin’s lymphoma (90Y-ibritumomab tiuxetan; Zevalin; IDEC Pharmaceuticals, San Diego, CA).
A number of 90Y-labeled peptides have been reported for PRRT, and 90Y labeled octreotide derivatives (targeting somatostatin (SST) receptor) are being extensively used for neuroendocrine tumor therapy (Bodei et al. 2004).
90Y labeled microspheres also are used for radioembolization purposes to treat hepatic cancer (Sangro et al. 2009). Microspheres having diameters larger than 10 μm accumulate in hepatic cancer after administration via the hepatic artery, the major supplier of blood to liver cancers, whereas the portal vein supplies most blood to normal liver tissue.
3.1.3 177Lu
177Lu is a relatively low-energy β-emitter that emits β-particles in three different maximum energy peaks (12 % 0.176 MeV, 9 % 0.384 MeV, and 79 % 0.497 MeV). It has a half-life of 6.75 days and a mean tissue penetrating depth of 0.28 mm. In addition, it emits γ-radiations of 113 keV (6.4 %) and 208 keV (11 %), which are useful for scintigraphic imaging. 177Lu is a reactor-produced radionuclide and is produced by irradiating enriched 176Lu in a reactor. 177Lu can be prepared in high yields at medium to high specific activity comparatively cheaply (Liu 2004). Until recently, 177Lu was only available from a reactor with a radioactive abundance of approximately 25 %. However, higher purity 177Lu (approximately 50 % pure) is now available, and anticipated improvements in the production and purification of 177Lu will allow further investigations to be conducted on its potential clinical utility (Michel et al. 2005a).
177Lu has a formal charge of +3 when complexed. The ionic radius of 177Lu (III) is 100.1 pm, and it is commonly complexed using DTPA and DOTA derivatives, such as 90Y, because 177Lu and lanthanide nuclides have chemical properties that resemble those of 90Y. In particular, lanthanides and 177Lu produce more stable complexes with DOTA than DTPA (Cacheris et al. 1987).
In terms of 177Lu applications, the focus of research has been on PRRT, and to date, several types of tumor-specific peptides, such as octreotide derivatives for SST receptor, Arg-Gly-Asp (RGD) peptide derivatives for αvβ3 integrin, and bombesin (BBN) analogs for gastrin-releasing peptide (GPR) receptor, have been reported (de Jong et al. 2001; Yoshimoto et al. 2008; Thomas et al. 2009).
The performances of 177Lu-based radioimmunotherapies have been compared with those based on 90Y (Vallabhajosula et al. 2005; Fortin et al. 2007). In a recent study, it was demonstrated that 177Lu-antibodies showed less non-specific cell killing than analogous antibodies labeled with 90Y in Raji B lymphoma cells. This result was attributed to the much higher energy of the 90Y β-particle (2.4 MeV) (Michel et al. 2005a).
177Lu has also been examined as a palliative radiotherapy for cancer bone metastasis. In a comparative study of 177Lu-EDTMP and 177Lu-DOTMP, 177Lu-EDTMP was found to accumulate in skeletal bone more so than 177Lu-DOTMP, whereas 177Lu-DOTMP was found to show lower retention in liver and kidneys and slightly greater blood clearance in animal models (Chakraborty et al. 2008).
3.1.4 186Re and 188Re
Rhenium has two candidate therapeutic radioisotopes (186Re and 188Re). 186Re emits β-radiation (Emax = 1.07 MeV, 91 %) and has a half-life of 3.68 days and a mean tissue penetration depth of 0.92 mm, and a γ-radiation (E = 137 keV, 9 %), which allows imaging during treatment. 186Re is produced in a reactor by irradiating 185Re with neutrons (185Re(n, γ)186Re). 186Re is not available with high specific activity, which limits its usefulness.
188Re was introduced relatively recently, and has excellent physical and chemical properties (Jeong and Chung 2003). 188Re emits high-energy β-radiation (Emax = 2.12 MeV, 85 %) with a half-life of 16.98 h and has a mean tissue penetration depth of 2.43 mm. In addition, it emits γ-radiation (155 keV, 15 %) which allows imaging during therapy, and it produces better image qualities than 186Re. 188Re can be prepared either by nuclear reaction (187Re(n, γ)188Re) or using a 188W–188Re generator. Generator-produced 188Re is carrier-free and has high specific activity. The major advantages of using 188Re in therapeutic nuclear medicine are that it is readily available and inexpensive, because it is produced using 188W–188Re generator which has a long shelf-life (longer than 4 months) due to the long half-life (69 days) of the 188W.
Rhenium and technetium are members of Group 7 of the periodic table, and therefore, have similar chemical properties. Accordingly, chelating agents suitable for technetium are also usually suitable for rhenium. However, the labeling conditions required, such as reaction temperatures, pH levels, and the amount of reducing agent, are often quite different. In general, rhenium requires higher reaction temperatures, lower pH levels, and higher concentrations of the stannous ion for reducing perrhenate than technetium.
Related posts:
 (Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
(Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
 Thyroid Carcinoma
Thyroid Carcinoma
 of Tumors: Pretargeting with Bispecific Antibodies
of Tumors: Pretargeting with Bispecific Antibodies
 Bisphosphonates for Bone Pain Palliation
Bisphosphonates for Bone Pain Palliation
 Radiopharmaceuticals for PET Dosimetry Before, During, and After Endoradiotherapy
Radiopharmaceuticals for PET Dosimetry Before, During, and After Endoradiotherapy
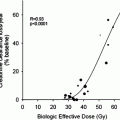 Use of Dosimetry in the Planning of Patient Therapy
Use of Dosimetry in the Planning of Patient Therapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


